Reviews in
Environmental Health, 1998
Environmental
Health Perspectives 106, Supplement 1, February 1998
Grantham's Landing, British Columbia, Canada
Key words: integrated defense system, multiple chemical sensitivity, disease model, IDS, MCS, traumatic dissociation, TD, pesticide, benzene, IL-1, vinyl chloride disease, autoimmune, overload, mercury allergy
Manuscript received at EHP 4 June 1997; accepted 9 October 1997.
Thanks to B. Bellin for research contributions and comments on the manuscript.
Address correspondence to Mr. S.C. Rowat, P.O. Box 175, 812 Port Mellon Highway, Grantham's Landing, British Columbia Canada V0N 1X0. Telephone: (604) 886-0670. Fax: (604) 886-0670. E-mail: steven_rowat@sunshine.net
Abbreviations used: ACTH, adrenocorticotropic hormone; APR, acute-phase response; BPD, borderline personality disorder; CNS, central nervous system; CRH, corticotropin-releasing hormone; d/p, direct or partial; EMU, environmental medical unit; GABA,-aminobutyric acid; GM-CSF, granulocyte-macrophage colony-stimulating factor; IDS, integrated defense system; Ig, immunoglobulin; IL, interleukin; IRA, immune response to antigen; MBP, mannan binding protein; MCS, multiple chemical sensitivity; MeHg, methyl mercury; MPD, multiple personality disorder; MT, metallothionen; NIR, nonspecific immune response; NK, natural killer; NS, neurogenic switching; OP, organophosphate; PCB, polychlorinated biphenyl; PTSD, post-traumatic stress disorder; SR, stress response; TD, traumatic dissociation; TDS, time-dependent sensitization; TNF-
, tumor necrosis factor alpha; VIP, vasoactive intestinal peptide.
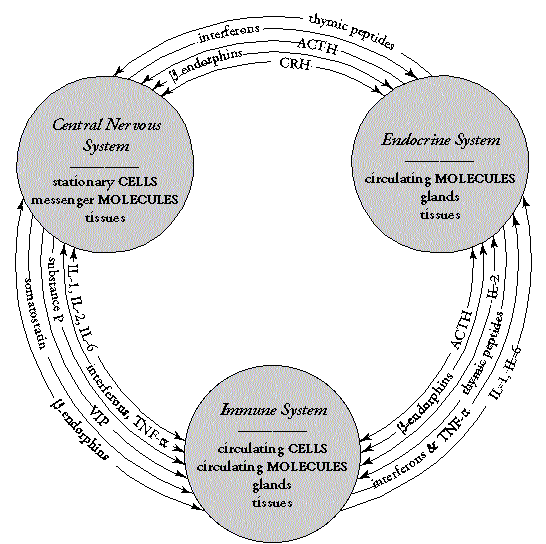
As Figure 1 illustrates neuroimmune-endocrine interactions of these
IDSs are complex. There are homeostases within each of the central nervous,
immune, and endocrine systems with multiple messengers running between
them. Many of these messengers travel between two sets of the systems such
as thymic peptides, adrenocorticotropic hormone (ACTH), tumor necrosis
factor alpha (TNF-![]() ),
and interleukins, or between all three such as ß-endorphins and interferons.
This overlap indicates not only that any one IDS is likely to invoke wide
systemic responses but also that overlaps and cross-interactions among
the IDSs themselves are possible.
),
and interleukins, or between all three such as ß-endorphins and interferons.
This overlap indicates not only that any one IDS is likely to invoke wide
systemic responses but also that overlaps and cross-interactions among
the IDSs themselves are possible.
 |
 |
|
| Figure 1. Messenger overlaps between the nervous, immune, and endocrine systems. Abbreviations: ACTH, adrenocorticotropic hormone; CRH, corticotropin-releasing hormone; TNF, tumor necrosis factor; VIP, vasoactive intestinal peptide. | Figure 2. Overlaps in range of symptomatic response of nine IDSs across the nervous, immune, and endocrine systems. |
If this overlap data is combined with the points listed above--the IDSs similarities in their range of symptomatic responses, the use of thresholds by the IDSs (some of which chain), long timeframes of certain IDSs, and conditioned learning, which cross-fertilizes inciters and triggering cues--there is potential for confounded activity among the IDSs that may include long-term learning of new thresholds of response. Such activity conceivably includes some existing diseases and syndromes. It may be especially useful to consider problematic diseases and syndromes like fibromyalgia, chronic fatigue, and multiple chemical sensitivity (MCS), which defy conventional analysis and often are postulated to be multifactorial conditions. Example models are generated for MCS in "MCS Mechanisms Generated from IDS Overlap Models."
Depending on the answers to these questions, we can build a combining hypothesis for the disease mechanism with several IDSs involved. This mechanism can include more than one type of IDS change. For instance (in a random example for illustration only): a) benzene can damage the immune response to antigen (19); b) continuous ambient air pollution such as NO2 produces a tolerance response, but with rising levels tolerance is eventually inadequate and is exhausted (20) and is followed by pathologic tissue changes; c) a child raised in an environment of severe abuse and neglect and without any caring adult support frequently will evolve a coping pattern that includes changes in, among other things, SR to aggressive situations, involving catecholamine, cortisol, serotonin, and endogenous opioid response changes (21).
If all three of these IDS changes were to occur in one person specific repeatable symptoms would be observed based on the interaction between the damaged IRA, the exhausted tolerance to NO2, and the modified (evolved) SR. If this specific constellation of IDS changes was to occur among a large number of people, reflecting a broad environmental context change in benzene concentration, NO2 concentration, and frequency of child abuse, then the resulting similarity of symptoms across separate patient case histories could be called a syndrome.
In this paper it is suggested that working backward from case histories using the IDS framework can in some instances reveal which IDS interactions cause the syndrome.
The full description of TD is included at the end of the listing below rather than in the Appendix, and readers are encouraged to examine it. The recent research into severe psychological trauma, including severe child abuse, has shown that it frequently produces permanent biological changes, and severe child abuse recently has been convincingly linked to well-known adult psychiatric dysfunctions. This background may be useful in understanding how TD could be an appreciable factor in widespread IDS interactions. The nine IDSs considered, along with referenced definitions and ranges of physical action, are listed below.
Time-Dependent Sensitization. "TDS refers to the ability of mild stressors--whether pharmacological or environmental--to induce physiological and behavioral effects which then progress, i.e., get stronger, entirely as a function of the passage of time since stressor presentation" (3).
"TDS is the progressive and persistent amplification of behavioral, neurochemical, endocrine, and/or immunological responses to repeated intermittent stimuli over time" (22).
The range of physical action includes neurotransmitters such as dopamine, norepinephrine, serotonin, GABA, and aspartate (3); hormones (corticosterone, ACTH, ß-endorphin); and immune system (3). Zinc is implicated in GABA (23-25) function.
Immune Response to Antigen. "The main feature that separates vertebrate from invertebrate immune systems is the ability to generate antigen-specific lymphoid cells" (26).
"[T]he immune system...learns to recognize foreign organisms and exhibits the property of memory. It remembers that it has seen a foreign organism and on its second encounter with that organism, it attacks more rapidly and more efficiently" (27).
The range of physical action for the IRA includes T- and B-cell receptors for specific antigens; thymus-directed T-cell growth processes including gene encoding for receptors (28); T- and B-cell multiplication; B-cell production of antigen-specific immunoglobulin antibody molecules including immunoglobulin (Ig)A, IgG, IgE, and IgM; T-cell production and reception of messengers including interleukins (IL-1 through IL-8). The range also includes TNF (2); platelet-activating factor (29); T- and B-cell production and reception of hormones including ACTH and ß-endorphin (30,31); T- and B-cell receptors for CNS messengers such as substance P (1,32), VIP (32,33), and somatostatin (32), and for endocrine hormones such as thymulin (34). Zinc is required for thymulin activation. For a review of dozens of messengers between the nervous and immune systems, see Plata-Salaman (29).
Kindling. "[K]indling refers to neural processes that mediate lasting changes in brain function in response to repeated, temporally spaced application of neurobehaviorally active agents" (35).
"Partial limbic kindling is a progressive and persistent lowering of the threshold for eliciting electrical after-discharges, but not motor seizures, in certain brain structures such as amygdala and hippocampus; behavioral consequences include increased avoidant behaviors" (22).
The range of physical action for kindling includes various brain structures, e.g., the cortex (36) and especially the limbic brain including the olfactory bulb and amygdala (37). Changes in brain chemistry are found, including a decrease in acetylcholinesterase enzyme activity that parallels the increase in sensitivity (37). Calcium-binding protein and tyrosine hydroxylase activity are reportedly reduced, and there are changes in ß-noradrenergic binding (36). Benzodiazepine receptor binding is modified (36), as are transmitter GABA and N-methyl-d-aspartate functions (4). Zinc may be implicated through GABA (23-25). Superoxide dismutase may also be involved (38). "These changes may be irreversible" (37).
Nonspecific Immune Response. "Long before the appearance of antigen-specific T cells or antibodies, the body is able to deploy an impressive array of humoral factors as a highly effective first line of defense against potential pathogens" (39).
The range of physical action for NIR includes circulating mast, natural
killer (NK), macrophage, B, and T cells and their produced molecules such
as TNF-![]() ,
IL-1 through IL-6, granulocyte-macrophage colony-stimulating factor (GM-CSF)
(40) and histamine (1). Other actions include collectins such as mannan
binding protein (MBP), surfactant protein-A, and surfactant protein-D (39);
and complement components such as C1r, C1s, C4, and C2 (39) which are serum
proteins that respond to antigen-antibody complexes (primarily) by generating
an enzyme cascade that effects the membrane attack complex for cytolysis.
,
IL-1 through IL-6, granulocyte-macrophage colony-stimulating factor (GM-CSF)
(40) and histamine (1). Other actions include collectins such as mannan
binding protein (MBP), surfactant protein-A, and surfactant protein-D (39);
and complement components such as C1r, C1s, C4, and C2 (39) which are serum
proteins that respond to antigen-antibody complexes (primarily) by generating
an enzyme cascade that effects the membrane attack complex for cytolysis.
Acute-phase Response. "In the aftermath of injury, trauma or infection of a tissue, a complex series of reactions are executed by the host in an effort to prevent ongoing tissue damage, isolate and destroy the infective organism and activate the repair processes that are necessary to return the organism to normal function" (41).
The range of physical action for APR includes circulating cells such
as macrophages, monocytes, platelets, and mast cells; stromal and endothelial
cells local to damage; cytokines such as IL-1, TNF, IL-6, and possibly
IL-4 and IL-8; thromboxane A2, and prostaglandins I2, E2, D2, and F2![]() .
Also included are leukotrienes B4, C4, D4, and E4, bradykinin; ACTH; glucocorticoids
including cortisol; ß-endorphin; Zn; MT; and many liver-mediated
proteins such as glycoprotein, C-reactive protein, complement C3, fibrinogen,
and antiproteases.
.
Also included are leukotrienes B4, C4, D4, and E4, bradykinin; ACTH; glucocorticoids
including cortisol; ß-endorphin; Zn; MT; and many liver-mediated
proteins such as glycoprotein, C-reactive protein, complement C3, fibrinogen,
and antiproteases.
Stress Response. "Stressors are the disturbing forces or threats to homeostasis, and adaptive responses include physical or mental reactions that attempt to counteract the effects of the stressors or disturbing forces in order to re-establish homeostasis" (6).
The range of physical action for SR includes brain regions: hypothalamus, brain stem, limbic system, and cortex; pituitary gland, adrenal gland, corticotropin-releasing hormone (CRH), ACTH, ß-endorphins, glucocorticoids, adrenergic system, and catecholamines. It also includes immune cells (macrophages, T, B, and NK cells); cytokines IL-1, IL-2, IL-3, IL-6, TNF, prostaglandins including PGE2; adenosine monophosphate (42); tissue Zn (43,44), and brain Zn (45).
Neurogenic Switching. "Neurogenic switching is proposed to result when a sensory impulse from a site of activation is rerouted via the central nervous system to a distant location to produce neurogenic inflammation at the second location" (46).
The range of physical action for NS includes immune system mast, T, B, and other cells; CNS peripheral cells; cytokines such as IL-1 and IL-2 communicating with both CNS and immune cells; neuropeptides such as substance P, somatostatin, and VIP communicating with both CNS and immune cells. Various regions of the central CNS may be involved in processing information during switching. Immune memory (cells and genetic coding) may also be involved in encoding the switching (10).
Tolerance. "Adaptation and tolerance are defined ultimately in biochemical and biological adjustments that enable organisms to survive...[T]olerance is characterized, not only by repair, but also by the development of refractoriness or immunity to the persistent insult so that repair at the [same] level is no longer required in the usual sense..." (20).
"Symptoms of exposure to many chemicals, whether inhaled or ingested, appear to follow a biphasic pattern. Adaptation is characterized by acclimatization (habituation, tolerance) with repeated exposures that result in a masking of symptoms" (47).
"The stage of resistance represents the sum of all nonspecific systemic reactions elicited by prolonged exposure to stimuli to which the organism has acquired adaptation as a result of continuous exposure. It is characterized by an increased resistance to the particular agent to which the body is exposed and a decreased resistance to other types of stress" (48).
"Oral tolerance is a term used to indicate antigen-specific systemic hyporesponsiveness after prior enteral (oral) exposure to that antigen" (49).
The range of physical action for tolerance includes multiple tolerance instances, mechanisms, and symptoms involving multiple organ systems (7,20,47-55); the entire nervous, immune, and endocrine systems are potentially involved.
Traumatic Dissociation. "Dissociation is defined in DSM-III-R [Diagnostic and Statistical Manual, 3rd Ed., Revised] as a disturbance or alteration in the normally integrative functions of identity, memory, or consciousness....a more precise definition of dissociation is required..." (56).
"Dissociation is one of the principal mechanisms by which people cope with overwhelming experiences. Dissociation terminates states of extreme physical and emotional arousal during the trauma, but over time dissociative processes may be activated by minor stresses and simple reminders of earlier trauma" (21).
"Dissociative states, used defensively at the time of abuse, may become a generalized defense in any situation where strong affect is aroused" (57).
"Dissociation may belong to the realm of biological mechanisms that are triggered by overwhelming trauma-related affects and events...." (58).
Current research and reviews assert that mild dissociation experiences are common in the general population (56) and that strong dissociation is a key and possibly defining component of post-traumatic stress disorder (PTSD) (21,59-62), borderline personality disorder (BPD) (57,58,60), and multiple personality disorder (MPD) cases (63). All these clinical psychopathologies are associated principally with childhood abuse (21,57-60,63-66) and their differences may represent stages and facets of the same underlying process (61,62,67,68).
Since the etiologies of PTSD, BPD, and MPD are only now being revealed and because they appear to overlap significantly (all three commonly involve child sexual abuse), TD as introduced here is modeled after dissociation as used by van der Kolk and Fisler (21,61,69) and others in which it refers to a combined biological-psychological mechanism central to all cases of PTSD, BPD, and MPD, and which also occurs in less extreme forms in nonpathologic populations.
TD, then, is an emergency defense system invoked during overwhelming inescapable psychological trauma, and modulates both cerebral logical organization and physiological affect regulation. Especially after chronic trauma in childhood, TD produces lasting changes in both, although the symptoms may not appear simultaneously.
MODE OF ACTION. Recent articles in attachment theory (59,70), a research and theoretical framework in developmental psychology, describe the building of a conceptual model of relationships in the infant mind as early as the end of the first year of life (70). Attachment researchers have identified several types of children with incomplete or deviant attachment behavior (called avoidant, resistant, and disorganized behavior). They have found these types of children to be the result of insensitive, inconsistent, and abusive parenting, respectively (59,70). These children are postulated to have developed incomplete or damaged internal models of themselves and their relationships to the outside world (70), possibly permanently, because abused children are known to more frequently become abusing parents (70).
Working from a framework of adult PTSD and BPD, van der Kolk and Fisler (21) postulated disrupted attachment (including that resulting from abuse) results in a biochemically based loss of conceptual control. Current perceptions that appear superficially similar to past trauma cause emotion-based fight or flight responses to occur immediately, without the mediation of an abstract internal model that could decide whether such responses are valid. van der Kolk and Fisler further maintain that "forty years of research on nonhuman primates have firmly established that...disruptions of attachment early in the life cycle (as occurs in child abuse) have long-term effects on the neurochemical response to subsequent stress, including the magnitude of the catecholamine response and the duration and extent of the cortisol response. There are also long-term effects on a number of other biological systems, such as the serotonin and endogenous opioid system" (21).
In humans as well, PTSD, BPD, and MPD have been associated with lasting biologic changes in either baseline levels or during response to stress. For PTSD measured in war veterans, this includes norepinephrine, cortisol, ACTH response to CRH, serotonin (71), endogenous opioids, lymphocyte glucocorticoid receptors (21,69), and regional brain-flow patterns (72). A serotonin function review has cited its low levels in BPD and shown low levels of serotonin to be related to hyperirritability, hyperexcitability, and hypersensitivity, pain sensitivity, and lack of an inhibitory response in behavioral systems subserved by the hippocampal/amygdala system, including conceptual processing of reward, punishment, and uncertainty (73).
MPD was only recently recognized as an intentionally secret condition and not as rare as had been thought (perhaps not rare at all). MPD may be a superordinate diagnosis that may contain any of the symptoms of PTSD, BPD, and other personality disorders (63). In MPD separate personalities may have different (allergic) immune reactions (65) as well as differing responses to the same substance (alcohol, foods, and medications) (65), different body temperature, heart rate, and skin temperature (66), different eye muscle malfunctions (visual acuity problems) (66) and insulin levels (74), different handednesses (65), and different brain wave patterns (66). Thus in extreme cases of TD (i.e., MPD), not only is there an altered response to stress, there is an altered psychologic and physiologic response to almost everything. The number of varying personalities, which can approach 200 (74), has been significantly connected (p<0.0001) to the number of types of childhood trauma reported in a patient's history (65).
Thus overwhelming trauma, particularly in inescapable (61,75) or double-bind situations [such as abuse by father or other primary caregiver (76)], appears to make a conceptual shift or dissociation in how both the psychologic and physiologic systems respond to stress. When the trauma occurs during the formation of a child's conceptual models, especially chronically, the shift is likely to be much more intense.
RANGE OF PHYSICAL ACTION. TD physical actions vary with the degree of dissociation; pathologically in PTSD and BPD they include many, and perhaps all, brain, endocrine, and immune systems that are involved in the SR such as cortisol, CRH, ACTH, norepinephrine, and lymphocytes; serotonin and possibly GABA and other parts of the noradrenergic system; and endogenous opioids. Although less well understood, TD also affects whatever higher-level brain centers subserve amnesia for self and place and for depersonalization and derealization experiences [such as not recognizing one's reflection in a mirror or looking at the world through a fog (56)]. In MPD the range of physical action includes all the foregoing plus whatever brain, immune, and endocrine areas and mediators are required to differentiate insulin levels, heart rate, skin temperature, allergic reactions, responses to foods and medications, eye acuity, handedness, and brain wave patterns. In other words, most if not all body systems are included.
INCITERS. TD inciters are primarily conceptual: inescapably traumatic (frightening) events; i.e., child physical and particularly sexual abuse, war, rape, torture, domestic violence (72), and natural disaster experiences (69).
Deciding who has MCS is problematic; the recently begun search for MCS biomarkers so far has been inconclusive (79-82). However, in the literature and based on extensive clinical experience, working definitions have evolved; this paper will use one similar to that proposed by Miller (83) and Miller et al. (84), with one notable difference: inclusion of the stress context.
Miller (83) proposes that MCS can be confirmed when, in a clean environmental medical unit (EMU), the following conditions are true: a) when a subject simultaneously avoids all chemical, food, and drug incitants, remission of symptoms occurs (unmasking); b) a specific constellation of symptoms occurs with reintroduction of a particular incitant; c) symptoms resolve when the incitant is again avoided; and d) with reexposure to the same incitant, the same constellation of symptoms reoccurs, provided the challenge is conducted within an appropriate window of time.
Working within the IDS overlap framework provided in "Integrated Defense System Overlaps as a Disease Model," a problem with this definition is that a particular form of psychological stress may be integral to the combined IDS response. For example, if TD has modified the SR and thereby modified the thresholds of other IDSs that use the same messengers (as modeled in the examples that follow), the above definition could lead to the improper attribution of symptoms to MCS if a concurrent psychological triggering of the SR is necessary for the low-level chemical response to occur. Because it is probable such a stress is not provided by the EMU, no response is found and MCS is therefore invalidated as a causative factor. Yet the response occurs again when the patient is in his or her normal (psychologically stressed) environment. Conversely, the patient may find the EMU stressful to the degree that it is not possible to unmask the symptoms. Consider the following report of a 21-year old patient with PTSD (whose response to chemicals is not reported): "A lot of times I wake up at two, three o'clock in the morning and I'm having an anxiety attack before my eyes even get open. Sometimes it's that I'm dreaming, because that [clinic visit] is still going around in my head, even though it was six months ago. I'm really scared of this one doctor, and a lot of times he's in [the dream]...I hyperventilate, and I can't breathe, and I have stomach spasms, muscle spasms, and sometimes I throw up after it's over" (85).
Bell et al. (86) expressed similar concern for MCS testing protocols because animal work shows TDS sensitization in one environment does not replicate in a novel environment. To put it simply: context matters.
Thus it may be that in searching for an MCS definition and mechanism the attempt to isolate the patient from the context is premature; patients' reports of what is happening to them in their own words may be our best source of data for formulating hypotheses of what is occurring. At present the only widely available forms of these reports are a few extremely moving self-reports that have appeared in the scientific literature (87,88) or the popular press and, importantly, collated data from clinicians (89-93), including attempts by clinicians to define the condition [collated by Miller (90)].
This wealth of rich, awkward, and unfinished data and other published reports, along with my own experiences with people reporting chemical sensitivity, have been used to generate the IDS-overlap MCS models to follow.
The overlap and chaining of thresholds in the examples developed below are suggested as first attempts at models that include nonlinear interactions between linked homeostatic systems. Mathematically they probably would best be expressed by a set of linked differential equations, although the explicit mathematics are not presented here. [Mathematics might best be supplied by chaos theory, which has been used to develop a model of the immune system as a dynamic adaptation system (27,55) to model the coevolution of hosts and parasites (97,98) and to study pathology (99), psychology (100), physiology (101), and a large number of other biomedical disciplines (102-107)].
Examples of the following MCS models produced through IDS interaction will be considered in the balance of this paper. Some are included because parts of their mechanism have been directly suggested by the MCS clinical literature; others are included because they are known to be widespread phenomena and their IDS interactions appear capable of generating MCS symptoms.
Six models are considered separately; they are grouped as follows:
| 1A. | Pesticide damage to the CNS |
| 1B. | Benzene disruption of IL-1 maturation |
| 1C. | Autoimmunity to IgG or vinyl chloride disease |
| 2A. | Chemical and stress overload (using a combination of formaldehyde, air pollutants, anxiety, benzene, alkylphenol novolac resin, and anger) |
| 2B. | Mercury allergy in a genetic subset (to increased levels of mercury in dental applications, food, and paint) |
| 3A. | MCS as evolution (a semantic exploration) |
Certain widely used pesticides that have disrupting effects on GABA neurotransmitters (4,75,113) may be initiators of kindling (4). This interference may also extend to polychlorinated biphenyls (PCBs) (75) and in general to organophosphate (OP) and organochlorine compounds (113). Some organochlorine insecticides such as chlorpyrifos reportedly cause both chronic neurologic symptoms (114) and modulation of the IRA (112). Certain OP insecticides enhance the large-fiber distal axonopathy caused by other OPs such as chlorpyrifos (109,114). Recently reported studies of Gulf War veterans confirm earlier reports of a connection between a syndrome of neurologic damage and wartime exposure to pesticides and insecticides (115-118).
Initial Damage. If we assume a relatively severe exposure to an OP such as chlorpyrifos with a coexposure to a synergistic damage-promoting solvent or other OP, there can be initial disruption of the GABA neurotransmission as well as damage to long nerve cells. It is also fair to assume that the nerve damage initially will invoke the NIR and, if the damage is sufficient, the APR.
Examining the IDS overlaps in Table 1, we see that the disruption of GABA may cause kindling and TDS to malfunction, as both use GABA. We also see that a large number of other messengers have been invoked by the NIR and the APR, including ILs, immune cells, ACTH, and ß-endorphin. ACTH and ß-endorphin, however, are also used by TDS. Because we are assuming that the GABA disruption also changed TDS, the situation is unstable. In one direction TDS may modify the APR through ACTH and ß-endorphin; in the other direction the APR may change TDS further, possibly including changing its threshold of reaction to incitants or the magnitude of its response. This bidirectional interaction is shown in Figure 3.
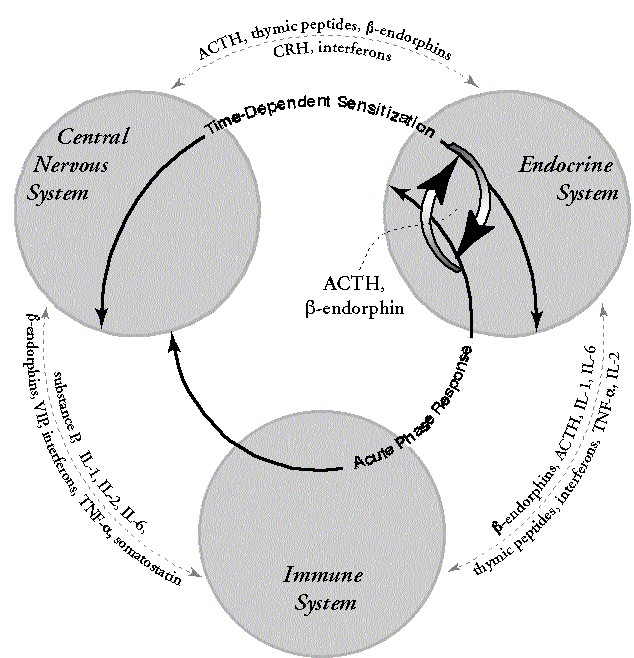
Figure 3. Modifications through overlap of messengers among IDSs. In multiple chemical sensitivity model 1A (pesticide damage to the CNS), TDS and APR are overlapping messengers ACTH, ß-endorphin, various hormones, metallothionein, zinc, and possibly others (Table 1). Such overlap provides opportunities for both intended and unintended bidirectional influences, including modification of triggering thresholds and changing the functions of messengers unique to each.
Extensions. With reference to Table 1, several major complications can be envisioned that would increase the chances of IDS interaction in this model; three will be discussed briefly.
BODY BURDENS AND SYNERGIES OF TOXIC CHEMICALS, INCLUDING PESTICIDES. Various chemical contaminants such as dioxins, PCBs, insecticides, and pesticides are reported in human tissue at levels that arouse concern (119-125). Because several chemicals are known to be present simultaneously in the normal body burden (119,125), synergies may occur. Examples are carbon tetrachloride with chlordecone (126-128) and chlorpyrifos with other OP pesticides (109,114). This could mean continuous disruption of GABA and unstable action of ACTH and ß-endorphin, possibly leading to unanticipated threshold changes in the TDS of various chemicals.
PREEXISITING TD. If a patient has a history of trauma sufficient to have modified his or her SR, psychological cues may incite changes in many messengers that also affect TDS, including dopamine and norepinephrine, ACTH, ß-endorphins, Zn, IL-1, and other interleukins, hormones, and cells. Such stress occurring concurrent with new chemical exposure could be the mechanism by which the response is spread (as explored in model 2A). At least one study has reported higher rates of self-reported childhood abuse among women reporting MCS than among controls (129), and it may be more than chance that a significantly higher proportion of females than males report MCS, as a similar female:male ratio occurs among those who are estimated to be sexually abused in childhood; about 4:1 in the former (130,131) and 3:1 in the latter (57).
EARLY AND ONGOING ZINC DEFICIENCY. Zinc deficiency has been widely investigated in the last two decades and is now recognized to be a public health problem (132); a recent estimate is that 30% of the adult U.S. population is at risk. Zinc is centrally involved in multiple human systems (133), including GABA and other neurotransmitter enabling (24,45,134-137), and in sequestering and defense against heavy metals and chemicals (138). Gestational Zn deficiency appears to damage hippocampal and other functions (44,45,139,140). Chronic Zn deficiency reduces the IRA by deactivation of thymulin (133,141-144). Thus several crossovers with TDS are possible. Limbic structures and GABA may be malfunctioning because of early Zn deficiency; or ongoing chronic Zn deficiency may, through lack of MT and increased infection and other malfunction of the immune response, modify TDS further, with crossovers at ILs, immunoglobulins, TNF, other immune cells, and ACTH and ß-endorphins.
Combining Extensions to Produce Variants of MCS. Given a base of a large number of people exposed to pesticides in our society, the logical combinations of these extensions with the initial exposure may generate symptomology, demographics, and incidence of MCS. That is, some exposed persons may already have body burdens of chemicals, some will have TD, and some will have Zn deficiency. Because all three conditions affect TDS, similar but not identical syndrome sets will be generated. Additionally some persons may have two of the three preconditions and a small set will have all three. These seven logical syndrome sets will share some characteristics but each will differ from the other six; each set will have a unique blend of incidence, intensity, and symptomology. A group of patients now clinically defined by one researcher as having MCS may constitute a particular skew of these seven sets, those defined by another a second skew, etc.
IL-1 holds a central place in neuroimmune-endocrine response. According
to reviews by Whitacre (2) and Plata-Salaman (29), IL-1 is produced by
many cell types, some immunologic, some endocrine, and some in the CNS.
IL-1 may be the central messenger in the SR (29), it has receptors in both
immune and CNS tissue (158), including the brain (158), and it can cross
the blood-brain barrier (159). Its effects include upregulation of IL-2,
interferon, and other lymphokines; it acts as a chemoattractant for T and
B cells, and it acts synergistically with IL-4 and IL-6. IL-1 also exerts
effects on NK cells, regulates cell growth including both immune and nerve
cells, promotes inflammation (prostaglandins), induces fever (160) and
slow-wave sleep, increases levels of epinephrine and vasoactive intestinal
peptide, induces synthesis and release of various pituitary hormones including
ACTH, and enhances the release of ß-endorphin, as well as having
other effects. Recent work on the evolutionary origins of IL-1 and the
other key cytokines IL-2 and TNF-![]() has shown them to be derived from invertebrate forms that perform similar
jobs (161), and in light of recent evidence of various immune (26,161)
and lectin (39,162-164) origins, it would appear that IL-1 holds central
place in the first memory system, the nonspecific immune system, which
predates even the existence of a CNS, much less a brain.
has shown them to be derived from invertebrate forms that perform similar
jobs (161), and in light of recent evidence of various immune (26,161)
and lectin (39,162-164) origins, it would appear that IL-1 holds central
place in the first memory system, the nonspecific immune system, which
predates even the existence of a CNS, much less a brain.
Along with the relatively new data about IL-1 suppression by benzene metabolites (19,145-148), benzene metabolites induce leukemia and other DNA damage (165-167). If this damage occurs at levels sufficient to invoke any of the eight IDSs besides kindling, IL-1 is likely to be involved as part of the IDS response; IL-1 is the single most shared messenger among the body's IDSs (Table 1).
One MCS study has shown IL-1 to be produced in significantly reduced amounts from peripheral blood mononuclear cells in cases relative to controls (81), although the authors state that this may have occurred because of laboratory methodology problems. Notwithstanding the slightness of this evidence, the IL-1-benzene connection appears to be a reasonable candidate for MCS investigation if the suppression of IL-1 maturation in bone marrow by benzene metabolites also holds in the large number of other sites where identical IL-1 is produced. It also may be noteworthy that the list of benzene-containing substances is close to being a list of the MCS substances most highly reacted to. If we suspect that which chemicals MCS sufferers react to may be a meaningful guide to what caused the condition, benzene is a good place at which to start investigating.
Initial Damage. If benzene metabolites have also downregulated the IL-1 an IDS is using for its messaging, the IDS response to benzene may be changed. For instance if we assume that benzene exposure induces the APR, then because of the benzene downregulation of IL-1, other messengers in the APR could have distorted values; these include ACTH, ß-endorphin, prostaglandins, other interleukins, mast cells, and other immune cells. TDS also uses ACTH and ß-endorphin; if it is invoked, its threshold of response may be modified. If the benzene exposure has been sufficient to induce the SR, then the distorted IL-1 values may additionally affect TDS through limbic brain structures and dopamine and norepinephrine, and kindling through these and through cortex and higher brain structures.
Extensions. CHILDHOOD WINDOW EXPOSURES. Childhood exposure data indicate that if exposure to benzene (or benzene-derived chemicals that have similar metabolites) occurred during developmental windows, special damage or immune learning could occur. Gestational windows correlate a wide variety of fetal malformations, toxicities, and subsequent developmental problems with various pregnancy exposures to PCBs (168), antibiotics (169,170), anesthetics (171-173), and environmental chemicals (174-177). Infant developmental windows have been reported to correlate hexachlorophene washing with neonatal brain damage and death (178,179). Such exposures of benzenoid chemicals may induce a TDS to benzene or its derivatives or an IRA to a hapten formed from such a chemical or its metabolites.
HAPTEN SPREADING FROM CONTINUOUS EXPOSURE. Antigenic haptens are formed from both aromatic and nonaromatic chemicals (180-184). An interesting series of experiments with acetaldehyde (183,185-189) has shown that the immune response successfully "...could be raised by immunizing with acetaldehyde conjugated to a carrier protein different from the one that is used for testing" (185). In other words the immune response could recognize the antigenic chemical segment and spread the reactivity to other carriers. In the case of acetaldehyde, usually IgE, and in one case IgG and IgM, is raised to acetaldehyde-protein adducts (183), and this has been used both in diagnosing alcoholism (186,187) and as an explanation for alcohol allergy (183). Benzene is the building block of the enormous number of new antibiotics, fuels, plastics, printing materials, food additives, pesticides, cleaners, and cosmetics that have been added to our environment in recent decades. If this immune recognition could happen with benzene metabolites or derivatives, the crossover between the IRA and another IDS, especially in light of the dysregulation of IL-1 by benzene, might account for both threshold response change and spreading found in MCS patients.
EARLY AND ONGOING ZINC DEFICIENCY. IL-1 and Zn-thymulin are synergistic. [Thymulin is central in T cell and other immune regulation (34) and Zn is required for thymulin function (142,190)]. IL-1 delivers Zn to thymulin via MT mRNA and Zn-thymulin potentiates IL-1 activation of nuclear protein kinase C in isolated splenocytes (191). Also, Zn deficiency in gestational and childhood windows, especially in conjunction with benzenoid environmental chemicals (192), reportedly causes a variety of malformations (44,193) including CNS development. Lack of Zn during early rat brain growth produces neurologic abnormalities (44,139,140). See additional discussion in section on model 1A.
BODY BURDENS AND SYNERGIES OF TOXIC CHEMICALS, INCLUDING PESTICIDES. Almost all the chemicals commonly in the body burden are benzenoid. See detailed description in section on model 1A.
PREEXISITING TD. In a human case-control study, psychological stress reduces the amount of IL-1 produced by peripheral blood leukocytes, which was correlated with the slowing of wound healing (194). See detailed description in section on model 1A.
Combining Extensions to Produce Variants of MCS. As discussed in the section on model 1A, these different situations can be combined to produce overlapping syndromes that could be subsets of MCS. Early window effects, hapten spreading, body burdens of benzenoid chemicals (and continuous exposure to benzene itself), preexisting TD, and Zn deficiency in early windows or later could interact individually with an IL-1 dysregulation model to increase or modify the effect. Any combination of two, three, four, or five of the extensions with the original could produce a different subset of symptoms and hence possibly appear as a different subset of the main syndrome.
Initial Damage. In our model we assume that chronic exposure to vinyl chloride has damaged both IgG and other tissues and the IRA produces T-cell-mediated autoimmune complexes raised against these tissues (196). We do not know with certainty the memory mechanism of such autoimmune disturbances; possibly it involves DNA encoding in the variable (diversity) joining recombination of T cells (26,28,201,202). In addition T memory cells exist for long periods, decades at least, without any booster from antigens (5,203).
Extensions. SPREADING TO OTHER IGG-INDUCING ANTIGENS. Certain foods such as bananas, milk, and eggs produce unusually high levels of IgG allergic responses in pooled human blood (199). Soy (204), beef (205), and corn (206) also induce IgG, as do various chemicals (181,182,206). Suppose a subject consumes one of these substances and has an IgG reaction to it; by increasing the IgG level, the subject is simultaneously increasing the total autoimmune reaction to IgG itself. This may induce an additional IDS such as NIR to become involved.
Because the NIR is also known to induce the immunoglobulins, it becomes possible that a feedback loop will be set up: with more IgG comes more IRA and more NIR; with more NIR comes more IgG. This escalation could be sudden in certain conditions (like a painful audio feedback), and, by multiple crossover messengers, might induce the SR and TDS to the initiating foods or chemicals. In this way, a large number of IgG-provoking substances eventually could become inciters of a TDS response.
SUDDEN SPREADING--MULTIPLE IDS SENSITIZATION TO IGG. This possibility, though apparently supported by no direct experimental evidence, produces such an interesting model that it must be at least mentioned briefly. What happens if after IgG becomes antigenic and IgG-inducing substances are introduced, a feedback loop causes TDS both to the inciting substance and to IgG? A spreading/multiplier effect is then set up. Any inciter of IgG automatically becomes an inciter also of TDS, even without a specific spreading event. The subject would suddenly become sensitized to all the foods and chemicals that already incite IgG, such as those mentioned in "Spreading to Other IgG-inducing Antigens." This type of quick spreading matches an important and puzzling aspect of the MCS syndrome.
FOOD-CONTAMINATING CHEMICALS. The presence of pesticide residues, additives, and packaging migrants in commercial foods provides an opportunity for chronic or acute conditioned association between a chemical and an IgG-inciting food. Additionally, if such a chemical became complexed with the food before or during metabolism, the association would be different and possibly more antigenic. It is perhaps noteworthy that such complexing does not appear to have been studied; the literature on food contaminants (surprisingly sparse) deals largely with a few identified migrants and contaminants (207-216) rather than with their ability to complex with foods. The intense European Economic Community initiative to develop measurement and reporting techniques for packaging migrants is based on specific lists of chemicals (217,218); unfortunately complexing with foods is not included.
Combining Extensions to Produce Variants of MCS. Reports of autoimmune markers in MCS patients (like most MCS data) are inconsistent. A recent report (89) shows antismooth muscle antibodies in about half the patients seen in a clinical practice but no data about anti-IgG antibodies or antibodies against any other IDS messenger. Although many MCS patients report food sensitivities, this model has been presented chiefly as illustration. It is plausible that any IDS messenger that could act as an antigen could provide a feedback mechanism for spreading of the MCS response. An MCS so produced could be viewed as a form of chemically formed autoimmune disease.
People who are middle-aged today were the first generation to have encountered these chemicals on a daily basis since birth. Because so few of these chemicals have had long-term testing, in a sense this generation is an experimental group. MCS is characterized by reactions to these chemicals, so it may be that MCS is the experimental result.
Initial Damage and Extensions. Model 2A will explore the possibility that specific configurations of increased multiple chemical exposure and stress can produce MCS through IDS interactions even though the IDSs are unchanged. In addition, Miller (90) and others (22,94) have developed models of MCS that propose sequential stages for MCS; common among these are susceptibility, sensitization, triggering, and spreading. For comparison's sake this description will adopt these stages; differences suggested by the IDS model will be discussed in "How Long is an Event?" Model 2A is shown graphically in Figure 4.
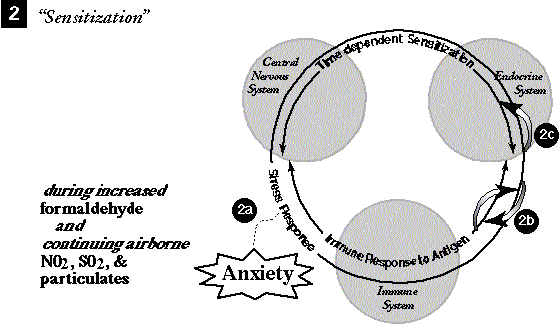
SUSCEPTIBILITY. As an example, suppose that a chronic low-level exposure (years long) to formaldehyde has created haptens and an IRA reaction in the subject (180-182). Assume that combined chronic chemical stressors such as SO2, NO2, and diesel exhaust particles are polluting the subject's work area simultaneously and also occurring in the subject's ambient urban air. These substances have impinged on the lung tissue (229) and a tolerance mechanism supplying compensatory lung tissue changes is occurring (20,48). Tolerance symptoms may not be noticed by the subject as a pattern because tolerance mechanisms are well known for moderating the magnitude of the reaction; in a sense, that is their function.
SENSITIZATION. A strong psychological anxiety (stress) occurs in combination with a moderately increased amount of formaldehyde. The anxiety induces the SR IDS. Because of the immune response already occurring and even increased slightly because of the increased level of formaldehyde, the crossover messengers between the SR and the immune response (for example, ACTH, ß-endorphins, IL-1 and -2, and immune cells, all of which are also involved in TDS) condition TDS to both formaldehyde and anxiety as inciters. Such bidirectional fertilization between psychological and chemical triggers is reported for TDS in animal experiments (3).
TRIGGERING. After some time the subject encounters a new chemical, benzene, while in a state of anxiety less severe than at the onset of the original event, so the SR IDS is not at first invoked. However TDS increases over time, even without exposures to the inciter; it is triggered by the anxiety. The tolerance mechanism, meanwhile, undisturbed during the sensitization stage, has continued to erode toward the exhaustion state (48). The added weight of the neurochemical and endocrine changes that characterize the TDS reaction causes the lung tolerance to air pollution to be breached and the NIR is invoked to deal with cellular damage that had been tolerated to that point. Under the influence of the concurrent benzene exposure, the APR is also cascaded from the NIR, and under the added influence of the TDS firing, the SR cascades. (Figure 4 shows a graphic separation of these events.) At this level of arousal TDS becomes conditioned to benzene also as a trigger, and we speculate that multiple overlaps in messengers between the TDS and the SR (Table 1) make it possible that the threshold for the SR becomes chained to the TDS response; in other words, the SR will be invoked directly by the TDS in future exposures.
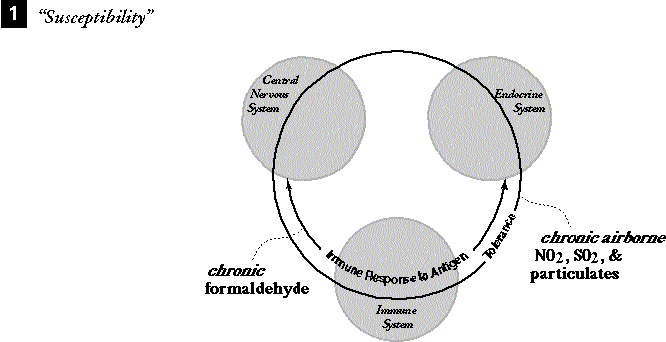 |
1. Chronic formaldehyde induces the immune response to antigen. Simultaneously, chronic SO2, NO2, and diesel and other particulate matter from air at the workplace and urban home impinge lung tissue and induce tolerance in the form of compensatory lung cell architecture. |
 |
2a. Acute anxiety occurs in combination
with moderately increased formaldehyde. The anxiety induces the SR.
2b. Crossover messengers interact between the SR and the IRA such as ACTH, ß-endorphins, IL-1 and IL-2, and immune cells. 2c. These messengers are also involved in TDS and TDS is invoked and conditioned to recognize both formaldehyde and anxiety as inciters. |
 |
3a. Later, subject encounters benzene
during moderate anxiety. TDS has increased over time and is triggered by
the anxiety.
3b. TDS reaction causes the already eroding lung tolerance to be breached and with continuing air pollution, the NIR is invoked to deal with cellular damage. 3c. Concurrent irritation/damage from the benzene exposure causes the APR to cascade from the NIR. 3d. Under the added influence of the TDS, the SR cascades. 3e. Multiple messenger overlaps chain the threshold for the SR to the TDS response so the SR will be invoked directly by the TDS in future. TDS also becomes conditioned to benzene as a trigger. |
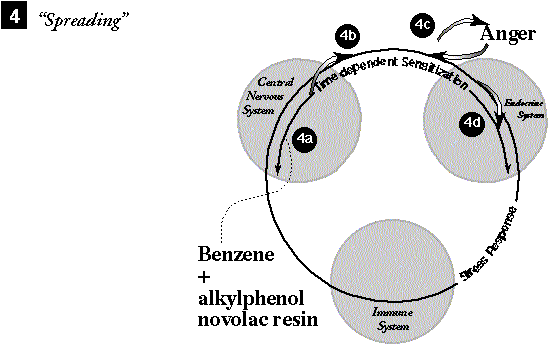 |
4a. Later, during moderate anger, benzene
is again encountered along with alkylphenol novolac resin used in no-carbon
forms. Benzene invokes TDS.
4b. TDS cascades the SR. 4c. Hormonal overlaps between the SR and anger increase anger's intensity. 4d. Messenger overlaps between increased anger, SR, and TDS cause TDS to become sensitized to alkylphenol novolac resin and anger as inciters. 4e. (Not shown.) In subsequent events this same spreading mechanism continues to add new stimulus chemicals and psychological stresses as TDS inciters. |
Figure 4. IDS interaction model 2A, of MCS resulting from chemical and stress overload, adapted from the necessary stages of MCS suggested by Miller (90) and others (22,94).
SPREADING. In a later event benzene is again encountered along with a new chemical, alkylphenol novolac resin, which is used in no-carbon-required forms (242) and a new psychological stress, moderate anger. Now benzene causes TDS and hence the SR to fire, and the anger increases in intensity. The TDS becomes conditioned to both the new chemical, alkylphenol novolac resin, and to anger, the new stress, as inciters.
In subsequent events this same spreading mechanism could continue to add new stimulus chemicals and psychological stresses and new symptoms at diverse sites.
Notes on the Model. In Figure 4, tolerance, which is no longer active, is not shown in stage three; nor is the IRA; in our example formaldehyde is no longer being encountered. Additional possibilities that could have been included are that the IRA could develop an associated specific reaction to benzene or its metabolites or that the formaldehyde chronic exposure could continue.
Interactions from Other Models. In particular, pesticide and other chemical body burdens under model 1A and interactions between benzene and IL-1, model 1B, may be of interest in this model. Childhood window exposures (model 1B) and TD (model 1A) could also be added. The multiple-feedback autoimmunity to IgG model (model 1C) could also be used, say, by substituting vinyl chloride for formaldehyde as the chronic exposure agent, but at this point the complications become mind-numbing.

Figure 5. Possible source of MCS biomarker data error. This graphic shows combined IDS overlap events in the level 3 triggering stage of chemical and stress overload MCS model 2A. If biochemical markers are taken indiscriminately within this period chaotic data may result, since the same neuroendocrine-immune messengers may be in use for different purposes at different times. (Figure 4 shows separated event sequence.)
Another difference is that in the current IDS model (model 2A) both sensitization and spreading are also occurring during this triggering sequence, since a new IDS is being added to the mechanism (SR) and a new inciter is being conditioned (benzene). During the nominal spreading stage, triggering of TDS and stress are also occurring. It appears that through the architecture of the IDS interaction and chaining models, which are composed of short discrete events of several types, various sequences and combinations of triggering, spreading, and sensitizing can be obtained.
These differences between the IDS model and the four-stage MCS model have testing implications that are discussed in "Relevance of the Models to Current MCS Research Recommendations."
In humans carrying dental amalgams, the amounts of Hg released have been estimated within the range that would cause reactions in those genetically susceptible to Hg (251). Therefore the millions of humans who carry Hg amalgams are theoretically at risk for genetically induced reactions. (Whether those not genetically susceptible could also be directly toxically affected by mercury is unknown; many widely divergent opinions and research reports have been published over the last few decades (247,248,251-256). Recent reports connect amalgams with oral cavity ill-health (248,257), increase in antibiotic-resistant bacteria in oral and intestinal cavities (258), multiple sclerosis (259,260), psychological complaints such as depression, excessive anger, and anxiety (261), and cardiovascular symptoms such as fatigue and high blood pressure (262).
Mercury is also available in the diet, particularly from fish. Levels of methyl mercury (MeHg) in fish appear to be growing and are a concern (263,264). An epidemiologic investigation of a fish-eating population is reported to show high susceptibility to brain damage during prenatal exposures to MeHg (263). Also Hg in paint is a widely recognized health hazard (265-267), as is skin-lightening cream (268). A syndrome of infant Hg toxicity called acrodynia is reported from application of calomel teething powders (255) and from paint exposures (267). A case-control study shows increased urinary Hg in people living in houses painted with Hg-latex paint (p<0.001) (267), and a case of acrodynia occurred in a child living in such a house (267). Before 1990 one-third of the interior latex paint used in the United States had added Hg fungicide (267).
Like MeHg, Hg is toxic to the CNS and established detrimental effects range from behavioral irritability and insomnia at low levels of exposure through tremors, muscle spasms, and nerve conduction loss at higher exposure levels (253). Many other toxic effects are reported (255), including cardiovascular (262), immune (248,255), and autoimmune effects (269).
Data on a direct connection of Hg exposure to MCS is lacking except for the synchronicity of neurologic symptoms, which are the most highly reported symptom category in MCS clinical literature and the symptoms most commonly produced by Hg. These reasons and the demographics prompted this exploratory model. Half the population of the western world has Hg amalgam fillings, and 5% may be allergic to Hg.
Initial Damage. We hypothesize a human strain that has a genetically determined immune response to Hg and a subject who has sufficient dental Hg released in the body to induce a systemic autoimmune reaction through the IRA. Subsequent to this the Hg level in the subject's body is being increased by food sources, further increasing the activity of the autoimmune response. Then a moderate-to-high exposure to toxic environmental Hg in paint, drugs, pesticides, cosmetics, or other source occurs during a stressful situation that includes a chemical exposure. For example Hg in fresh paint, in cosmetics or calomel, or in amalgam implantation in a dentist's office in conjunction with anesthetic injection or ambient chemicals might provide this combination. This exposure causes SR activation, and in combination with the ongoing autoimmunity, a combined CNS-mediated TDS to Hg and to the stress-associated chemical may begin. Because there is already an appreciable body burden of Hg and also an immune response to this Hg, there may be a messenger crossover--ACTH, ß-endorphin, IL-1, or other immune cells or messengers, for example--between the TDS and the immune and SRs. This may change the threshold of response of any of these IDSs. This combined sensitization of the immune response and TDS to internal Hg may create an unstable situation that allows subsequent exposures to become conditioned inciters of the TDS during various situations; e.g., those that elicit movement or increase of the Hg body burden while stressing psychologically or chemically.
Extensions. HEAVY METALS/ZINC DISPLACEMENT. Cd, Pb, and Hg are all Zn agonists, and rising levels of Cd and Pb can add to the toxic effect of Hg in the body. Metals are unique in the environment in that they are not consumed by human enterprise and so continue to increase in the biosphere as we collect and process them (270); one review concludes that we have a growing problem of heavy metal toxicity (270). Cd is increasing in human tissue. It has a human half-life of 30 years (271) and the average human body burden since the turn of the century has increased by a factor of 4.7 according to one study (272), with the renal concentration increasing by a factor of 47 (272). Another study found a renal increase factor of 3.8 (273). Cd is a potent developmental toxin in animal studies; its effects include CNS and behavioral dysfunctions and many others (274). Some Cd effects have been linked to its displacement of Zn from various enzymes (270,274), and Cd from maternal smoking is thought to decrease the transfer of Zn across the human placenta (275,276), which may implicate Cd in multiple teratogenic effects strongly suspected to result from Zn gestational deficiency (44,193). Pb caused a reduction in cognitive development through 7 years of age in children living in a lead-smelting community (277). Pb is also reported to cause DNA-protein crosslinks and DNA repair inhibition (270), and to accumulate in the hippocampus of children, rats, and monkeys. In monkeys the effects are accompanied by Zn displacement and learning deficits (278).
CHILDHOOD OR GESTATIONAL MERCURY EXPOSURES. MeHg exposure during pregnancy causes neurologic abnormalities, including psychomotor deficits, mental retardation, and deafness in children (279). Fetal effects occur at much lower exposure levels than required for effects in adults, although in both cases effects are almost exclusively on the nervous system, especially the CNS (268). Hg is not known to damage the fetus as heavily as MeHg, but this may be because of lack of study (253).
Combining Extensions to Produce Variants of MCS. Body burdens of chemicals, preexisting TD, early and ongoing Zn deficiency described under model 1A, and childhood window exposures and hapten spreading described under models 1B and 1C could also interact in this model. Again various subsets of the MCS syndrome might be produced depending on which of these substrates is present.
Concluding Note on Inadequate or Inappropriate IDS Models 2A and 2B. In these two thought experiments, physiologically healthy MCS subjects appear plausible through various interactions of the nine IDSs. That is, it may be that in some forms of MCS the subjects' neuroimmune-endocrine systems are functioning well--within the limits of their present design. These design limitations may now be coming to our attention because of interactions with increasing concentrations of new types of toxic chemicals and metals and possibly new levels of stress. In this model those who have MCS are at the end of a normal distribution; i.e., they have been unlucky in how many chemicals and how much stress and heavy metal they were exposed to or in what synergistic combinations. They may have marginally different thresholds to TDS, kindling, or other IDSs than the norm. A disturbing correlate of these models is that an increasing population in the industrial world may be at risk for developing MCS.
It appears that the neuroendocrine-immune system is purposely unique in each individual, and part of this comes from experience. Jerne (7) suggests that "Early imprints leave the deepest traces."
If we consider the crossover messengers (Table 1) used by most IDS--messengers such as IL-1, ACTH, and ß-endorphin, MT, mast cells, etc.--limited resources the body allocates between IDS in times of attack, we may, in a semantic reorientation, be able to model IDS threshold changes as a learning effect rather than inadequacy or damage. It would be reasonable also if the body set these allocation ranges in advance; that may be one function of childhood windows and of neuroimmune learning in general. For instance TD can produce MPD alternative personalities that have differing immune responses (65) and many other lifelong physiologic differences; the appropriate alternative personality appears in a specific context then disappears when the context changes. Various combinations of IDSs therefore may be interacting meaningfully to adjust thresholds in readiness for special situations.
Childhood developmental windows could be understood then not only as times when toxicants can accidentally produce more damage but also as specific times when the IDSs are sampling the external environment to determine a range of response appropriate for the lifetime of a particular individual.
In a species sense MCS may conceivably constitute a healthy response to an unhealthy environment. Although MCS symptoms frequently are painful, pain can have a useful function. That is, since the chemicals MCS individuals avoid are known to be toxic in high concentrations and are also increasingly recognized as toxic and carcinogenic in chronic exposures (239,285), (Table 2), such a difference could be viewed as an improvement in the species. People who get MCS would be subject to lower rates of chemical damage and cancer because they would have an increased impetus to avoid the toxicants.
In such a model we consider it possible that it is through such IDS learning mechanisms that people with MCS have learned to abandon almost all aspects of 20th-century life that involve the smell, touch, and taste of rubbers and synthetic foams, new plastics, fresh paints, petrochemical cleaners, oil and gas fuels, and petroleum-based pesticides, perfumes, food additives, and drugs; also that their inability to be near these chemicals is probably lifelong (89). We may even imagine that persons with MCS may find themselves living in an electrically heated rugless house in a small village far from a city, with a yard bordering a wild field. When they sit in the yard on a sunny day breathing good air, listening to the birds chirping, watching their organic garden growing, and looking at the car parked in the driveway, which they can only use once a week for a 20-min trip, it may occur to them that their MCS disease--as a learning experience--might not be such a bad thing in some ways.
Relevance of the IDS Models to Current MCS Research Recommendations. If biomarker data are pursued and the interactive models developed in this paper are even partially on the right track, complex nonlinear biomarker data will be required. But how do we even know who has MCS? It has been calculated that if only 10% of a study population are responders, then to achieve a response shift of one standard deviation from norm, with p=0.01 90% of the time, the study population would need to be 2102 people, but only 23 people if 100% are responders (291). Thus, defining a small population of people with MCS in the absence of reliable biomarkers is a catch-22. Perhaps long-term self-control crossover studies (291,292) that can measure data at various points in the triggering-spreading sequence would be more useful, but the number of such studies that could be economically carried out in a limited timeframe is small. Also there is no way to know if we are measuring people with MCS or people who are willing to spend time in a physician's office.
Recent research groups working on MCS testing (84,293) recommend using only people who have had a verifiable single-exposure event precipitating their MCS. But how representative of the entire population is this? Not only do we not know how many gradual onset cases are being left out, the crossover with chronic fatigue and fibromyalgia, among other conditions, is reportedly high. These conditions may even be indistinguishable in most cases (89). If it turns out that an IDS model accounting for all these effects simultaneously is required--perhaps what Miller has called "an emerging category of disease" (294)--then limiting studies to only a small number of adult single-event precipitated MCS cases in a clinical setting may seriously decrease our knowledge base for building the pattern.
An additional serious problem with clinical testing discussed in "Working Definition" is that some of the IDS models developed in this paper predict that people with MCS will have psychological reactions in a testing situation, which will distort results. Conversely they may require a particular type of psychological stress to produce their reaction, and such stress may only occur when they are in their ordinary life settings. Bell et al. (86), in their working-group report on MCS testing, referred to the need to avoid novelty in testing situations (a variant of this same problem) and concluded, "it will be essential to perform multiple, not one exposure sessions separated in time....That is, it is necessary to initiate and elicit sensitization within the same experiment." This is an important limitation and may be difficult to achieve. Cohen et al. (293), in another working group, state the problem clearly: "Do events perceived as stressful in the recent or past history of the individual play roles in the onset and/or progression of MCS? ...Which factor is more important in the traumatic initiation: the exposure to chemicals associated with the traumatic event, and/or the stressful experience associated with the event? Can a stressor precipitate MCS in a chemically sensitized individual who is not displaying overt symptoms of MCS at the time of stressor exposure?"
Yet their recommendations on the design of controlled exposure studies (293), according to my interpretation, do not circumvent this. In making recommendations on testing three other working groups from the same conference (77,84,295) made no reference to this problem, yet it seems crucial.
Animal studies may help clarify the situation. The Bell et al. (86) working group suggests a series of experiments on rodents with each series taking measurements from microelectrode bundles in the brain. The series roughly measures: a) chemical odor only, b) stress first, then chemical odor, and c) chemical odor first, then stress. This allows the use of stress, which the human model does not. It does, however, leave out variables explored in some of the IDS models; for example, it has no simultaneous stresses and chemicals and no provision for combinations of early developmental windows, body burdens of chemicals and metals, gestational and chronic Zn deficiency, or a genetic allergy. These variables could be added in future experiments, but finding the right combination(s) might be time consuming and possibly even prohibitive in nonhuman subjects.
Also there is the problem outlined in "How Long is an Event? Comparisons of IDS Models with the Four-stage Model of MCS" that several very short IDS-chaining events may be being lumped together under what is nominally called sensitization, triggering, or spreading. Additionally, these events may constitute combinations of sensitization, triggering, and spreading and even more importantly may use identical messengers that perform changing functions within very short time spans. Unless the measuring protocol is programmed for such shifts, chaotic biomarker data could result.
Rigorous double-blind (84) and balanced-placebo design (295) clinical testing suggested by some work groups may be critical in convincing other investigators, doctors, and legislators of the legitimacy of MCS as a debilitating condition, and should probably be undertaken immediately. But these tests may not identify the origins of MCS. An analogy is that for a century it has been possible to view the symptoms of MPD or BPD in clinical settings and the disorders have long been accepted as valid. Yet only recently has a convincing etiology been discovered (57,58,60,65,66); this has been through collating large numbers of patient questionnaire self-reports. These early traumas were not available in clinical settings to either the people who experienced them or the health care workers who had not.
Identifying MCS Mechanisms by Large-scale Self-report Epidemiology. An alternative investigation route that appears logical is large-scale self-report epidemiology of MCS. After decades of argumentative discourse among the medical profession and the press, people with MCS are aware and beginning to get organized, even if they do not congregate in cities or at conferences. They act most frequently through newsletters and memberships in local self-help organizations. There are many national organizations and newsletters, especially when the associated conditions of fibromyalgia and chronic fatigue are considered. It is possible that by using e-mail and the telephone, 10 organizations could quickly be found that would each provide addresses for 500 persons. Within a short time, a mailing could be sent to 5000 people, all of whom had self-defined long-term MCS. This mailing could be done either at the same time as or in advance of animal and clinical studies called for by the research groups working on MCS.
One suggestion might be for each member of a panel of six to eight people who have been studying this condition for many years to produce 50 to 100 questions based on his or her work. These questions would then be collected, sorted, and culled to a workable number--perhaps 150 questions. These questions would be mailed, returned, and statistically analyzed for useful patterns that might suggest directions for future testing and research.
It appears appropriate to suggest that at least one panel member prepare questions based on the IDS interaction models. It also seems reasonable to have at least one panel member with overt MCS symptoms and who has lived with MCS for an appreciable period of time. This might expand the functional perspective of the panel; and help validate the undertaking for those approached to contribute addresses and contacts as well as those asked to complete the questionnaire. It would also be important that at least one panel member have previous experience with wide-area questionnaire preparation, distribution, and analysis. Finally, including some carefully designed open-ended and single- and multiple-choice types of questions might allow more robust cross-checking of the data. This has recently been demonstrated in a long-term questionnaire study searching for developmental correlates in an adult population (296).
Several nonexclusive mechanisms of MCS based on these IDS models appear plausible. If one or more of them are accurate, demonstrating the existence of MCS and verifying its mechanisms may involve a simultaneous study of clinical, animal, and large-scale self-report data.
In brief the specific immune response has the important characteristics of long-term memory through memory cells and probably genetic coding; tolerance through suppressor T cells that modulate other immune functions; and associative spreading of the response by recognition of multiple epitopes on one antigen, and possibly by other mechanisms such as CNS-mediated conditioned learning.
T lymphocytes are activated by antigen presented to them by macrophages or other presenter cells, and in turn they grow, multiply clonally, and begin expressing molecular messengers that in turn activate B cells, other T cells, and other types of cells to aid in attack and other responses to that specific antigen (2). Many of these messengers communicate directly with receptors in the endocrine organs or on CNS cells (32), so the response can involve all body systems even if the antigen is local. The immune responses can in turn be modified by input from the CNS and endocrine messengers. In addition an autoimmune T-cell-mediated response is sometimes mounted against the host's own tissue. This can occur when the tissue is complexed with a foreign antigen or for other unknown reasons.
A major discovery of recent years is that antigen-specific memory T cells, which will precipitate a faster, stronger response on second encounter with the antigen than naive T cells (203) and B cells, can sometimes persist for a lifetime (203) without any intervening booster encounters with the original antigen (5,299).
Another important recent discovery is that the antigen-recognizing T-cell receptors undergo directed gene rearrangement early in T-cell growth in the thymus (26,28). Thus, it appears possible that antigen recognition is being genetically encoded (201,202), although such a feedback loop has not been characterized in detail.
Locally a second wave of cytokines is released by stromal cells, and local endothelial cells undergo changes that induce leukocytes into the tissue. An arachidonic acid cascade occurs, including prostaglandins and thromboxanes, and vascular tone changes occur, swelling and redness, for example. Histamine, serotonin, and platelet-activating factor change tissue flow of fluids. Pain occurs through molecules such as bradykinin, which are released during clotting (41).
Distantly the interleukins and TNF can have multiple effects (29,158,288), including changing the hypothalamic temperature setting (fever) and inducing changes in liver metabolism. Distinct sets of acute-phase proteins are induced separately in the liver by IL-1 as opposed to IL-6-type cytokines (41); the former include glycoprotein, C-reactive protein and complement C3, the latter include fibrinogen and antiproteases (41,288). Liver changes also include production of MT and the consequent lowering of plasma Zn levels (44,138,301).
These cytokines can also induce ACTH from the adrenal-pituitary axis, generating corticosteroids that feed back to suppress the immune reactions (6,41); this can be accompanied by direct production of analgesic ß-endorphins by B cells (31). At this point the CNS is directly involved, as CRH can be induced in the brain by interleukins and psychological stress (281), and it induces ACTH and hence corticosteroids. This description merges with that of the SR described below.
Although it may be that the APR diminishes by an undirected erosion over a 24- to 48-hr period, it is also reported that IL-4 and IL-10, produced by some T cells, can accomplish downregulation of IL-1, IL-6, and IL-8 (41).
With central perception of stress, several feedback loops are invoked within the neuroendocrine-immune system; the interactions between these, although complex, presumably determine how the response occurs, its intensity, and how long it lasts (31,281).
The main functional changes are energy redistribution toward improved attention and alertness and suppression of less essential behavior such as feeding and sexual interest (6). The brain, certain muscles, and other organs of immediate need are supplied with more oxygen and nutrients (6).
The immune response is also significantly suppressed at various levels (6). This has been well documented (29,32,281,303) and includes cells (macrophage, T, B, NK, etc.) and downregulation of cytokines such as IL-1, IL-2, and others (304-306). Less research reports differential effects, with some immune parameters showing increased activity (303); this may be time dependent (305).
The main effectors are corticosteroids and catecholamines; the former have been more extensively documented and are presumed to be centrally induced, although this may prove not to be true with the advent of newer evidence. CRH, induced in the brain by stressors (including interleukins) induces ACTH from the pituitary and thus corticotropin from the adrenal gland. CRH is associated with SR behaviors such as anorexia, decreased libido, and motor activity changes. Fear responses and decrease in willingness to explore have been reported at higher dosages (6).
CRH also induces--and is induced by--the central autonomic arousal system (norepinephrine), resulting in simultaneous expression of catecholamines, which contribute, with the glucocorticoids, to both behavioral and immune effects.
On the one hand mediators such as IL-1 and IL-2 participating in IgE and other immune responses also affect the release of neuropeptides such as substance P from nearby nerve cells. On the other hand, substance P, a known participant in neurogenic inflammation caused by chemicals, also can incite the IgE response and cause other immune changes. Thus a feedback loop can be initiated.
Although not using the term NS, another review (32) has shown how this could happen in the intestines, where neuropeptides substance P, somatostatin, and VIP are produced by nerve cells, received by various immune cells, and produced by various immune cells.
Because we know the CNS can be conditioned to control immune responses (10), for instance to have an audiovisual signal control IgE response to egg albumin in the intestines (13), and that immune responses can quickly become systemic reactions that include CNS modulation (such as in anaphylactic shock after insect bite), this feedback loop of cross-communication between adjacent nerve and immune cells has been suggested to be a likely candidate for explaining such puzzling conditions as migraine, asthma, arthritis, and MCS (46).
Because an overall mechanism for the induction of tolerance has yet to be postulated, we remain at the descriptive stage. However tolerance is so widely reported in human and other life-form responses to various insults that it may be useful to define it as being any back-up system developed by an organism that allows it to more successfully bear a chronic injury.
Tolerance eventually gives out. In other words all of the forms of tolerance described below--with the possible exception of certain forms of nonpathogenic food tolerance (49)--delay or minimize damage but eventually succumb to a long-term chronic stressor. During this process of succumbing (exhaustion) (48) systemic changes occur; e.g., rat lungs may lose tolerance to NO2. Stephens et al. (20) describe this process as follows "...pathogenesis continued to produce additional alterations at various biological levels, first stepwise and then simultaneously, and so become identified at some point as a 'clinical entity'...It appears that adaptation may not be sustained in the face of persistent stimulation from an injurious agent...."
Reported examples of tolerance include changes in Escherichia coli colony-forming when treated with Cd (309); in rat lung tissue repair in response to NO2 (20) and gasoline (310); in human psychophysiologic responses such as body balance and reaction time to inhaled xylene (311), ozone, and various solvents (53); and in response syndromes to Zn oxide fumes, nitroglycerin, and cotton and other grain dusts (47). One review of examples (47) imputed to tolerance and its interruption tells of: brain- and CNS-mediated behavioral effects; asthma; an infectionlike syndrome that includes fever, coughing, nausea, and weakness; a syndrome with drowsiness, severe headaches, and vomiting that sometimes includes violent behavior; a syndrome with airway irritation, coughing, pulmonary function test reductions and red blood cell fragility; emphysemalike changes and fibrosis; olfactory fatigue; and excess stimulation and tiredness in an alternating pattern (indicating repeated tolerance interruption).
A special type of tolerance has been proposed for the immune system; various cells such as T and B cells are widely believed to function by suppression (7,55) and anergy (54), which are forms of self-controlled tolerance. Yet another form, food tolerance or intolerance, also involves the immune system, including the immuo-globulins, but may be distinct since it has some unusual features (49). Addiction, including food addiction (312), has been described as an unfortunate type of tolerance (47,52); in this case, the organism procures and self-administers the insulting agent to enjoy certain aspects of the tolerance mechanism (52) or to ensure that tolerance continues (47,312).
Masking and masked addiction are terms applied to the inability of a self-conscious organism to know the inciting agent (78). It occurs during tolerance when the exposures are continuous and the symptoms are diffuse. It can occur for foods (312) and it has been proposed that masked tolerance to household chemicals is a significant unrecognized health problem (313).
At least one tolerance mechanism is described as proceeding by adaptive changes in DNA transcription (309). Possibly the recent change of scientific paradigm (still under debate) that allows for directed genetic adaptation (28,202) as opposed to merely random mutation will stimulate our understanding of other DNA mechanisms of tolerance if these exist.
Tolerance mechanisms have been suggested as an underappreciated reason why dose-response curves are frequently nonlinear (51).
References
1. Payan DG. Substance P: a modulator of neuroendocrine-immune function. Hosp Pract (February 15):67-80 (1989).
2. Whitacre CC. Immunology: a state of the art lecture. Ann NY Acad Sci 594:1-16 (1990).
3. Antelman SM. Time-dependent sensitization in animals: a possible model of multiple chemical sensitivity in humans. Toxicol Ind Health 10(4-5):335-342 (1994).
4. Adamec R. Modelling anxiety disorders following chemical exposures. Toxicol Ind Health 10(4-5):391-420 (1994).
5. Lau LL, Jamieson BD, Somasundaram T, Ahmed R. Cytotoxic T-cell memory without antigen. Nature 369(23 June):648-652 (1994).
6. Chrousos GP. Molecular and endocrine mechanisms of the stress response, pp 854-857. In: Sternberg EM, Moderator, The Stress Response and the Regulation of Inflammatory Disease. Ann Intern Med 117(10):854-866 (1992).
7. Jerne NK. Towards a network theory of the immune system. Ann Immunol 125(C):373-389 (1974).
8. Biondi M. The application of the human stress model to psychoneuroimmunology. Acta Neurol 13(4):328-334 (1991).
9. Ader R, Cohen N. Conditioned immunopharmacologic effects on cell-mediated immunity. Int J Immunopharmacol 14(3):323-327 (1992).
10. Grossman Z, Herberman RB, Livnat S. Neural modulation of immunity: conditioning phenomena and the adaptability of lymphoid cells. Int J Neurosci 64(1-4):275-290 (1992).
11. Ader R, Kelly K, Moynihan JA, Grota LJ, Cohen N. Conditioned enhancement of antibody production using antigen as the unconditioned stimulus. Brain Behav Immun 7(4):334-343 (1993).
12. Olness K, Ader R. Conditioning as an adjunct in the pharmacotherapy of lupus erythematosus. J Dev Behav Pediatr 13(2):124-125 (1992).
13. MacQueen G, Marshall J, Perdue M, Siegel S, Bienenstock J. Pavlovian conditioning of rat mucosal mast cells to secrete rat mast cell protease 2. Science 243:83-84 (1989).
14. Workman EA, La Via MF. T-lymphocyte polyclonal proliferation: effects of stress and stress response style on medical students taking national board examinations. Clin Immunol Immunopathol 43(3):308-313 (1987).
15. Ader R, Cohen N. Psychoneuroimmunology: conditioning and stress [Abstract]. Annu Rev Psychol 44:53-85 (1993).
16. La Via MF, Workman EA. Psychoneuroimmunology: where are we, where are we going? Recenti Prog Med 82(12):637-641 (1991).
17. Kim JJ, Fanselow MS. Modality-specific retrograde amnesia of fear. Science 256(5057):675-677 (1992).
18. Bolla-Wilson K, Wilson RJ, Bleecker ML. Conditioning of physical symptoms after neurotoxic exposure. J Occup Med 30(9):684-686 (1988).
19. Renz JF, Kalf GF. Role for interleukin-1 (IL-1) in benzene-induced hematotoxicity: inhibition of conversion of pre-IL-1 alpha to mature cytokine in murine macrophages by hydroquinone and prevention of benzene-induced hematotoxicity in mice by IL-1 alpha. Blood 78(4):938-944 (1991).
20. Stephens RJ, Freeman G, Evans MJ. Early response of lungs to low levels of nitrogen dioxide. Arch Environ Health 24(March):160-179 (1972).
21. van der Kolk BA, Fisler RE. Childhood abuse and neglect and loss of self-regulation. Bull Menninger Clin 58(2):145-168 (1994).
22. Bell IR. White paper: neuropsychiatric aspects of sensitivity to low-level chemicals: a neural sensitization model. Toxicol Ind Health 10(4-5):277-312 (1994).
23. Vautrin J, Schaffner AE, Barker JL. Tonic GABA secretion of cultured rat hippocampal neurons rapidly transformed by Zn2+ into quantal release. Neurosci Lett 158(2):125-129 (1993).
24. Xie XM, Smart TG. A physiological role for endogenous zinc in rat hippocampal synaptic neurotransmission. Nature 349(6309):521-524 (1991).
25. Xie X, Smart TG. Giant GABAB-mediated synaptic potentials induced by zinc in the rat hippocampus: paradoxical effects of zinc on the GABAB receptor. Eur J Neurosci 5(5):430-436 (1993).
26. Thompson C. New insights into V(D)J recombination and its role in the evolution of the immune system. Immunity 3(5):531-539 (1995).
27. Farmer JD, Kauffman SA, Packard NH, Perelson AS. Adaptive dynamic networks as models for the immune system and autocatalytic sets. Ann NY Acad Sci 504:118-131 (1987).
28. Gill JI, Gulley ML. Immunoglobulin and T-cell receptor gene rearrangement. Hematol Oncol Clin North Am 8(4):751-770 (1994).
29. Plata-Salaman CR. Immunoregulators in the nervous system. Neurosci Biobehav Rev 15(2):185-215 (1991).
30. Ballieux RE. Bidirectional communication between the brain and the immune system. Eur J Clin Invest 1:6-9 (1992).
31. Kavelaars A, Ballieux RE, Heijnen CJ. The role of interleukin-1 in the CRF- and AVP-induced secretion of ir-B-endorphin by human peripheral blood mononuclear cells. J Immunol 142:2338-2342 (1989).
32. Ottaway CA. Neuroimmunomodulation in the intestinal mucosa. Gastroenterol Clin North Am 20(3):511-529 (1991).
33. Wenger GD, D'Orisio SM, Goetzl EJ. Vasoactive intestinal peptide: messenger in a neuroimmune axis. Ann NY Acad Sci 594:104-119 (1990).
34. Millington G, Buckingham JC. Thymic peptides and neuroendocrine-immune communication. J Endocrinol 133(2):163-168 (1992).
35. Adamec RE, Stark-Adamec C. Limbic hyperfunction, limbic epilepsy, and interictal behavior: models and methods of detection. In: The Limbic System: Functional Organization and Clinical Disorders (Doane BK, Livingston KE, eds). New York:Raven, 1986;129-145.
36. Burnham WM, Niznik HB, Kish SJK. Biochemical changes in the limbic system following kindling: assay studies. In: The Limbic System: Functional Organization and Clinical Disorders (Doane BK, Livingston KE, eds). New York:Raven, 1986;123-127.
37. Girgis M. Biochemical patterns in limbic system circuitry: biochemical-electrophysiological interactions displayed by chemitrode techniques. In: The Limbic System: Functional Organization and Clinical Disorders (Doane BK, Livingston KE, eds). New York:Raven, 1986;55-65.
38. Mori N, Wada JA, Watanabe M, Kumashiro H. Increased activity of superoxide dismutase in kindled brain and suppression of kindled seizure following intra-amygdaloid injection of superoxide dismutase in rats. Brain Res 557(1-2):313-315 (1991).
39. Holmskov U, Malhotra R, Sim RB, Jensenius JC. Collectins: collagenous C-type lectins of the innate immune defense system. Immunol Today 15(2):67-74 (1994).
40. Ansel JC, Brown JR, Payan DG, Brown MA. Substance P selectively activates TNF-alpha gene expression in murine mast cells. J Immunol 150(10):4478-4485 (1993).
41. Baumann H, Gauldie J. The acute-phase response. Immunol Today 15(2):74-80 (1994).
42. Glaser R, Kennedy S, Lafuse WP. Psychological stress-induced modulation of interleukin 2 receptor gene expression and interleukin 2 production in peripheral blood leukocytes. Arch Gen Psych 47:707-712 (1990).
43. Arizono K, Tanahe A, Ariyoshi T, Moriyama M. Induction of metallothionein by emotional stress. In: Metallothionein in Biology and Medicine (Klaassen CD, Suzuki KT, eds). Boca Raton, FL:CRC Press, 1991;271-282.
44. Keen CL, Taubeneck MW, Daston GP, Rogers JM, Gershwin ME. Primary and secondary zinc deficiency as factors underlying abnormal CNS development. Ann NY Acad Sci 678:37-47 (1993).
45. Itoh T, Saito T, Fujimura M, Watanabe S, Saito K. Restraint stress-induced changes in endogenous zinc release from the rat hippocampus. Brain Res 618(2):318-322 (1993).
46. Meggs WJ. Neurogenic switching: a hypothesis for a mechanism for shifting the site of inflammation in allergy and chemical sensitivity [Abstract]. Environ Health Perspect 103(1):54-56 (1995).
47. Ashford A, Miller CS. Key terms and concepts. In: Chemical Exposures: Low Levels and High Stakes. New York:Van Nostrand Reinhold, 1991;27-58.
48. Selye H. The general adaptation syndrome and the diseases of adaptation. J Allergy 17:231-247, 289-323, 358-398 (1946).
49. Strobel S. Dietary manipulation and induction of tolerance. J Pediatr 121(5 Pt 2):S74-S79 (1992).
50. Randolph TG, Moss RW. The ups and downs of addicted life. In: An Alternative Approach to Allergies. New York:Harper & Row, 1980;36-48.
51. Davis JM, Svendsgaard DJ. U-shaped dose-response curves: their occurrence and implications for risk assessment. J Toxicol Environ Health 30(2):71-83 (1990).
52. Newlin DB. Drug sensitization, substance abuse, and chemical sensitivity. Toxicol Ind Health 10(4-5):463-480 (1994).
53. Miller CS. Possible models for multiple chemical sensitivity: conceptual issues and role of the limbic system. Toxicol Ind Health 8(4):181-202 (1992).
54. Bemer V, Rovira P, Truffa-Bachi P. T-cell activation, anergy and immunomodulation by molecules of viral, fungal and vegetal origin. Res Immunol 146(4-5):249-262 (1995).
55. Farmer JD, Packard NH, Perelson AS. The immune system, adaptation, and machine learning. Physica 22D:187-204 (1986).
56. Ross CA, Joshi S, Currie R. Dissociative experiences in the general population: a factor analysis. Hosp Community Psychiatry 42:297-301 (1991).
57. Ogata SN, Silk KR, Goodrich S. Childhood sexual and physical abuse in adult patients with borderline personality disorder. Am J Psychiatry 147:1008-1013 (1990).
58. Saunders EA, Arnold F. A critique of conceptual and treatment approaches to borderline psychopathology in light of findings about childhood abuse. Psychiatry 56(2):188-203 (1993).
59. Alexander PC. Application of attachment theory to the study of sexual abuse. J Consult Clin Psychol 60(2):185-195 (1992).
60. Saxe GN, van der Kolk BA, Berkowitz R, Chinman G, Hall K, Lieberg G, Schwartz J. Dissociative disorders in psychiatric inpatients [see comments] [Abstract]. Am J Psychiatry 150(7):1037-1042 (1993).
61. van der Kolk BA, Fisler R. Dissociation and the fragmentary nature of traumatic memories: overview and exploratory study [Abstract]. J Trauma Stress 8(4):505-525 (1995).
62. van der Kolk BA, Herron N, Hostetler A. The history of trauma in psychiatry [Abstract]. Psychiatr Clin North Am 17(3):583-600 (1994).
63. Kluft RP. Clinical presentations of multiple personality disorder. Psychiatr Clin North Am 14(3):605-629 (1991).
64. Barthauer L. A piece of my mind: secret pain. Primary Care 20(2):xvii-xix (1993).
65. Putnam FW, Guroff JJ, Silberman EK. The clinical phenomenology of multiple personality disorder: a review of 100 recent cases. J Clin Psychiatry 47:285-293 (1986).
66. Ross CA. Epidemiology of multiple personality disorder and dissociation. Psychiatric Clin N Am 14(3):503-517 (1991).
67. Divac-Jovanovic M, Svrakic D, Lecic-Tosevski D. Personality disorders: model for conceptual approach and classification. Part I: general model. Am J Psychother 47(4):558-571 (1993).
68. Lonie I. Borderline disorder and post-traumatic stress disorder: an equivalence? Aust NZ J Psychiatry 27(2):233-245 (1993).
69. van der Kolk BA, Fisler RE. The biologic basis of posttraumatic stress. Primary Care 20(2):417-432 (1993).
70. Zeanah CH, Zeanah PD. Intergenerational transmission of maltreatment: insights from attachment theory and research. Psychiatry 52(2):177-196 (1989).
71. van der Kolk BA, Dreyfuss D, Michaels M, Shera D, Berkowitz R, Fisler R, Saxe G. Fluoxetine in posttraumatic stress disorder [see comments] [Abstract]. J Clin Psychiatry 55(12):517-522 (1994).
72. Rauch SL, van der Kolk BA, Fisler RE, Alpert NM, Orr SP, Savage C R, Fischman AJ, Jenike MA, Pitman. RK. A symptom provocation study of posttraumatic stress disorder using positron emission tomography and script-driven imagery. Arch Gen Psychiatry 53(5):380-387 (1996).
73. Depue RA, Spoont MR. Conceptualizing a serotonin trait. A behavioral dimension of constraint. Ann NY Acad Sci 487:47-62 (1986).
74. Blume ES. Multiple personality disorder. In: Secret Survivors: Uncovering Incest and its Aftereffects in Women. New York:Ballantine, 1990;86-88.
75. Corrigan FM, Coulter F. Neuroleptic malignant syndrome, amitriptyline, and thioridazine [Letter]. Biol Psychiatry 23(3):320-321 (1988).
76. Herman J, Russel D, Trocki K. Long-term effects of incestuous abuse in childhood. Am J Psychiatry 143:1293-1296 (1986).
77. Bascom R, Meggs WJ, Frampton M, Hudnell K, Killburn K, Kobal K, Medinsky M, Rea W. Neurogenic inflammation: with additional discussion of central and perceptual integration of nonneurogenic inflammation. Environ Health Perspect 105(Suppl 2):531-537 (1997).
78. Ashford A, Miller CS. Chemical exposures and sensitive populations. In: Chemical Exposures: Low Levels and High Stakes. New York:Van Nostrand Reinhold, 1991;3-26.
79. Subcommittee on Immunotoxicology. Use of biologic markers in controversial areas of environmental health. In: Biologic Markers in Immunotoxicology. Washington:National Academy Press, 1992;127-148.
80. Albright JF, Goldstein RA. Is there evidence of an immunologic basis for multiple chemical sensitivity? Toxicol Ind Health 8(4):215-219 (1992).
81. Simon GE, Daniell W, Stockbridge H, Claypoole K, Rosenstock L. Immunologic, psychological, and neuropsychological factors in multiple chemical sensitivity: a controlled study. Ann Intern Med 119(2):97-103 (1993).
82. Broder I. Chairman's Introductory Remarks: Environmental Sensitivities Workshop. Chron Dis Canada Suppl(Environmental Sensitivities Workshop):6 (1991).
83. Miller CS. Toxicant-induced loss of tolerance--an emerging theory of disease? Environ Health Perspect 105(Suppl 2):445-453 (1997).
84. Miller CS, Ashford N, Doty R, Lamielle M, Otto D, Rahill A, Wallace L. Empirical approaches for the investigation of toxicant-induced loss of tolerance. Environ Health Perspect 105(Suppl 2):515-519 (1997).
85. Gottleib A. Post traumatic stress disorder and vulvar pain. Vulvar Pain Newslett 8(Fall):1-15 (1995).
86. Bell IR, Rossi J, Gilbert ME, Kobal G, Morrow LA, Newlin DB, Sorg BA, Wood RW. Testing the neural sensitization and kindling hypothesis for illness from low levels environmental chemicals. Environ Health Perspect 105(Suppl 2):539-547 (1997).
87. Keplinger H. Patient statement: chemically sensitive. Toxicol Ind Health 10(4-5):313-317 (1994).
88. Wilson C. Patient statement: chemical sensitivity--one victim's perspective. Toxicol Ind Health 10(4-5):319-321 (1994).
89. Ziem G, McTamney J. Profile of patients with chemical injury and sensitivity. Environ Health Perspect 105(Suppl 2):417-436 (1997).
90. Miller CS. White paper: chemical sensitivity: history and phenomenology. Toxicol Ind Health 10(4-5):253-276 (1994).
91. Miller CS, Mitzel HC. Chemical sensitivity attributed to pesticide exposure versus remodeling. Arch Environ Health 50(2):119-29 (1995).
92. Bell IR, Peterson JM, Schwartz GE. Medical histories and psychological profiles of middle-aged women with and without self-reported illness from environmental chemicals. J Clin Psychiatry 56(4):151-160 (1995).
93. Bell IR, Schwartz GE, Peterson JM, Amend D. Self-reported illness from chemical odors in young adults without clinical syndromes or occupational exposures. Arch Environ Health 48(1):6-13 (1993).
94. Ashford A, Miller CS. Mechanisms of multiple chemical sensitivities. In: Chemical Exposures: Low Levels and High Stakes. New York:Van Nostrand Reinhold, 1991;88.
95. Vojdani A, Ghoneum M, Brautbar N. Immune alteration associated with exposure to toxic chemicals. Toxicol Ind Health 8(5):239-254 (1992).
96. Simon TR, Hickey DC, Fincher CE, Johnson AR, Ross GH, Rea WJ. Single photon emission computed tomography of the brain in patients with chemical sensitivities. Toxicol Ind Health 10(4-5):573-577 (1994).
97. Anderson RM, May RM. Coevolution of hosts and parasites. Parasitology 85(Pt 2):411-26 (1982).
98. May RM. Parasitic infections as regulators of animal populations. Am Sci 71:36-45 (1983).
99. Cross SS, Cotton DW. Chaos and antichaos in pathology [Abstract]. Hum Pathol 25(7):630-637 (1994).
100. Barton S. Chaos, self-organization, and psychology [Abstract]. Am Psychol 49(1):5-14 (1994).
101. Elbert T, Ray WJ, Kowalik ZJ, Skinner JE, Graf KE, Birbaumer N. Chaos and physiology: deterministic chaos in excitable cell assemblies [Abstract]. Physiol Rev 74(1):1-47 (1994).
102. Goldsmith JR, Kordysh E. Why dose-response relationships are often non-linear and some consequences. J Exposure Anal Environ Epidemiol 3(3):259-276 (1993).
103. Butz M. Practical applications from chaos theory to the psychotherapeutic process, a basic consideration of dynamics. Psychol Rep 73(2):543-554 (1993).
104. Reidbord SP, Redington DJ. Psychophysiological processes during insight-oriented therapy: further investigations into nonlinear psychodynamics. J Nerv Ment Dis 180:649-657 (1992).
105. Gleick J. Chaos: Making A New Science. New York:Penguin, 1987;352.
106. Bassingthwaighte JB. Chaos in cardiac signals [Abstract]. Adv Exp Med Biol 346:207-218 (1993).
107. Preisler H, Raza A. An overview of some studies of chronic myelogenous leukemia: biological-clinical observations and viewing the disease as a chaotic system [Abstract]. Leuk Lymphoma 11(Suppl 1):145-150 (1993).
108. Rosenthal NE, Cameron CL. Exaggerated sensitivity to an organophosphate pesticide [Letter]. Am J Psychiatry 148(2):270 (1991).
109. Moretto A, Lotti M. Promotion of peripheral axonopathies by certain esterase inhibitors. Toxicol Ind Health 9(6):1037-1046 (1993).
110. Lamielle M. Indoor air: Congress and flight attendant address polluted cabin air. Delicate Balance 5(3-4):7-8 (1992).
111. Cone JE, Sult TA. Acquired intolerance to solvents following pesticide/solvent exposure in a building: a new group of workers at risk for multiple chemical sensitivities? Toxicol Ind Health 8:29-39 (1992).
112. Thrasher JD, Madison R, Broughton A. Immunologic abnormalities in humans exposed to chlorpyrifos: preliminary observations. Arch Environ Health 48(2):89-93 (1993).
113. Corrigan FM, MacDonald S, Brown A, Armstrong K, Armstrong EM. Neurasthenic fatigue, chemical sensitivity and GABAa receptor toxins [Abstract]. Med Hypotheses 43(4):195-200 (1994).
114. Kaplan JG, Kessler J, Rosenberg N, Pack D, Schaumburg HH. Sensory neuropathy associated with Dursban (chlorpyrifos) exposure. Neurology 43(11):2193-2196 (1993).
115. Haley RW, Horn J, Roland PS, Bryan WW, Van Ness PC, Bonte FJ, Devous MD Sr, Mathews D, Fleckenstein JL, Wians FH Jr et al. Evaluation of neurologic function in Gulf War veterans: a blinded case-control study [Abstract]. JAMA 277(3):223-230 (1997).
116. Haley RW, Kurt TL. Self-reported exposure to neurotoxic chemical combinations in the Gulf War: a cross-sectional epidemiologic study [Abstract]. JAMA 277(3):231-237 (1997).
117. Haley RW, Horn J, Kurt TL. Is there a Gulf War Syndrome? Searching for syndromes by factor analysis of symptoms [Abstract]. JAMA 277(3):215-222 (1997).
118. The Iowa Persian Gulf Study Group. Self-reported illness and health status among Gulf War veterans [Abstract]. JAMA 277(3):238-245 (1997).
119. Kutz FW, Wood PH, Bottimore DP. Organochlorine pesticides and polychlorinated biphenyls in human adipose tissue. Rev Environ Contam Toxicol 120:1-82 (1991).
120. Unger M, Olsen J. Organochlorine compounds in the adipose tissue of deceased people with and without cancer. Environ Res 23:257-263 (1980).
121. Jensen GE, Clausen J. Organochlorine compounds in adipose tissue of Greenlanders and Southern Danes. J Toxicol Environ Health 5:617-629 (1979).
122. Wolff MS, Anderson HA, Rosenman KD, Selikoff IJ. Equilibrium of polybrominated biphenyl (PBB) residues in serum and fat of Michigan residents. Bull Environ Contam Toxicol 21:775-781 (1979).
123. Muhlebach S, Wyss PA, Bickel MH. The use of 2,4,5,2´,4´,5´-hexachlorobiphenyl (6-CB) as an unmetabolizable lipophilic model compound. Pharmacol Toxicol 69(6):410-415 (1991).
124. Lisk DJ. Environmental implications of incineration of municipal solid waste and ash disposal. Sci Total Environ 74:39-66 (1988).
125. Kutz FW, Cook BT, Carter-Pokras OD, Brody D, Murphy RS. Selected pesticide residues and metabolites in urine from a survey of the U.S. general population. J Toxicol Environ Health 37(2):277-291 (1992).
126. Calabrese EJ, Baldwin LA, Mehendale HM. G2 subpopulation in rat liver induced into mitosis by low-level exposure to carbon tetrachloride: an adaptive response. Toxicol Appl Pharmacol 121(1):1-7 (1993).
127. Rao SB, Mehendale HM. Halomethane-chlordecone (CD) interactive hepatotoxicity--current concepts on the mechanism. Indian J Biochem Biophys 30(4):191-198 (1993).
128. Mehendale HM. Potentiation of halomethane hepatotoxicity by chlordecone: a hypothesis for the mechanism. Med Hypotheses 33(4):289-299 (1990).
129. Staudenmayer H, Selner ME, Selner JC. Adult sequelae of childhood abuse presenting as environmental illness [Abstract]. Ann Allergy 71(6):538-546 (1993).
130. Fiedler N, Kipen HM. Chemical sensitivity: the scientific literature. Environ Health Perspect 105(Suppl 2):409-415 (1997).
131. Gibson PR. Environmental illness/multiple chemical sensitivities: invisible disabilities. Women Ther 14(3/4):171-185 (1993).
132. Sandstead HH. Is zinc deficiency a public health problem? Nutrition 11(Suppl 1):87-92 (1995).
133. Walsh CT, Sandstead HH, Prasad AS, Newberne PM, Fraker PJ. Zinc: health effects and research priorities for the 1990s. Environ Health Perspect 102(Suppl 2):4-46 (1994).
134. Slevin JT, Kasarskis EJ. Effects of zinc on markers of glutamate and aspartate neurotransmission in rat hippocampus. Brain Res 334:281-286 (1985).
135. Harrison NL, Radke HK, Talukder G, Ffrench-Mullen JM. Zinc modulates transient outward current gating in hippocampal neurons. Recept Channels 1(2):153-163 (1993).
136. Moynahan EJ. Zinc deficiency and disturbance of mood and visual behaviour. Lancet (January 10):91 (1976).
137. Frederickson CJ, Danscher G. Zinc-containing neurons in hippocampus and related CNS structures. Prog Brain Res 83:71-84 (1990).
138. Sato M, Bremner I. Oxygen free radicals and metallothionein. Free Radic Biol Med 14(3):325-337 (1993).
139. Dreosti IE, Manuel SJ, Buckley RA, Fraser FJ, Record IR. The effect of late prenatal and/or early postnatal zinc deficiency on the development and some biochemical aspects of the cerebellum and hippocampus in rats. Life Sci 28:2133-2141 (1981).
140. Crawford IL, Connor JD. Zinc in maturing rat brain: hippocampal concentration and localization. J Neurochem 19:1451-1458 (1972).
141. Allen JI, Kay NE, McClain CJ. Severe zinc deficiency in humans: association with a reversible T-lymphocyte dysfunction. Ann Intern Med 95:154-157 (1981).
142. Dardenne M, Bach J. Rationale for the mechanism of zinc interaction in the immune system. In: Nutrient Modulation of the Immune Response (Cunningham-Rundles S, ed). New York:Marcel Dekker, 1993;501-509.
143. Dardenne M, Pléau JM, Savino W, Prasad AS, Bach JF. Biochemical and biological aspects of the interaction between thymulin and zinc. Prog Clin Biol Res 380:23-32 (1993).
144. Fraker PJ, DePasquale-Jardieu P, Zwickl CM, Luecke RW. Regeneration of T-cell helper function in zinc-deficient adult mice. Proc Natl Acad Sci USA 75(11):5660-5664 (1978).
145. Kalf GF, Renz JF, Niculescu R. p-Benzoquinone, a reactive metabolite of benzene, prevents the processing of pre-interleukins-1 alpha and -1 beta to active cytokines by inhibition of the processing enzymes, calpain, and interleukin-1 beta converting enzyme [Abstract]. Environ Health Perspect 104(Suppl 6):1251-1256 (1996).
146. Niculescu R, Bradford HN, Colman RW, Kalf GF. Inhibition of the conversion of pre-interleukins-1 alpha and -1 beta to mature cytokines by p-benzoquinone, a metabolite of benzene [Abstract]. Chem Biol Interact 98(3):211-222 (1995).
147. Niculescu R, Kalf GF. A morphological analysis of the short-term effects of benzene on the development of the hematological cells in the bone marrow of mice and the effects of interleukin-1 alpha on the process [Abstract]. Arch Toxicol 69(3):141-148 (1995).
148. Miller AC, Schattenberg DG, Malkinson AM, Ross D. Decreased content of the IL-1 alpha processing enzyme calpain in murine bone marrow-derived macrophages after treatment with the benzene metabolite hydroquinone [Abstract]. Toxicol Lett 74(2):177-184 (1994).
149. Mehlman MA. Dangerous and cancer-causing properties of products and chemicals in the oil refining and petrochemical industry. VIII: Health effects of motor fuels: carcinogenicity of gasoline--scientific update. Environ Res 59(1):238-249 (1992).
150. Litovitz T. Myocardial sensitization following inhalation abuse of hydrocarbons. Occup Med 3(3):567-568 (1988).
151. Reid WD, Ilett KF, Glick JM, Krishna G. Metabolism and binding of aromatic hydrocarbons in the lung: relationship to experimental bronchiolar necrosis. Am Rev Respir Dis 107:539-551 (1973).
152. Autrup H, Jeffrey AM, Harris CC. Metabolism of benzo[a]pyrene in cultured human bronchus, trachea, colon, and esophagus. In: Polynuclear Aromatic Hydrocarbons: Chemistry and Biological Effects, Fourth International Symposium. Columbus, OH:Battelle Press, 1977;89-105.
153. Wolff SP. Correlation between car ownership and leukaemia: is non-occupational exposure to benzene from petrol and motor vehicle exhaust a causative factor in leukaemia and lymphoma? Experientia 48(3):301-304 (1992).
154. Scheepers PT, Bos RP. Combustion of diesel fuel from a toxicological perspective. II: Toxicity. Int Arch Occup Environ Health 64(3):163-177 (1992).
155. Yardley-Jones A, Anderson D, Parke DV. The toxicity of benzene and its metabolism and molecular pathology in human risk assessment. Br J Ind Med 48(7):437-444 (1991).
156. MacFarland HN. Toxicology of petroleum hydrocarbons. Occup Med 3(3):445-454 (1988).
157. Nesnow S, Evans C, Stead A, Creason J, Slaga TJ, Triplett LL. Skin carcinogenesis studies of emission extracts. In: Toxicological Effects of Emissions From Diesel Engines (Lewtas J, ed). New York:Elsevier Biomedical, 1981;295-320.
158. Cunningham E Jr, De Souza EB. Interleukin 1 receptors in the brain and endocrine tissues. Immunol Today 14(4):171-176 (1993).
159. Banks WA, Kastin AJ, Durham DA. Bidirectional transport of interleukin-1 alpha across the blood brain-barrier. Brain Res Bull 23:433-437 (1989).
160. Rothwell NJ. CNS regulation of thermogenesis [Abstract]. Crit Rev Neurobiol 8(1-2):1-10 (1994).
161. Cooper EL, Rinkevich B, Uhlenbruck G, Valembois P. Invertebrate immunity: another viewpoint. Scand J Immunol 35:247-266 (1992).
162. Licastro F, Davis LJ, Morini MC. Lectins and superantigens: membrane interactions of these compounds with T lymphocytes affect immune responses. Int J Biochem 25(6):845-852 (1993).
163. Vasta GR, Ahmed H, Fink NE, Elola MT, Marsh AG, Snowden A, Odom EW. Animal lectins as self/non-self recognition molecules. Biochemical and genetic approaches to understanding their biological roles and evolution. Ann NY Acad Sci 712:55-73 (1994).
164. McCoy JJ, Mann BJ, Petri WA Jr. Adherence and cytotoxicity of Entamoeba histolytica or how lectins let parasites stick around. Infect Immun 62(8):3045-3050 (1994).
165. Eastmond DA. Induction of micronuclei and aneuploidy by the quinone-forming agents benzene and O-phenylphenol [Abstract]. Toxicol Lett 67(1-3):105-18 (1993).
166. Robertson ML, Eastmond DA, Smith MT. Two benzene metabolites, catechol and hydroquinone, produce a synergistic induction of micronuclei and toxicity in cultured human lymphocytes [Abstract]. Mutat Res 249(1):201-209 (1991).
167. Kolachana P, Subrahmanyam VV, Meyer KB, Zhang L, Smith MT. Benzene and its phenolic metabolites produce oxidative DNA damage in HL60 cells in vitro and in the bone marrow in vivo [Abstract]. Cancer Res 53(5):1023-1026 (1993).
168. Jacobson JL, Jacobson SW, Humphrey HE. Effects of in utero exposure to polychlorinated biphenyls and related contaminants on cognitive functioning in young children. J Pediatr 116(1):38-45 (1990).
169. Schwarz RH. Considerations of antibiotic therapy during pregnancy. Obstet Gynecol 58(Suppl 5):95S-99S (1981).
170. Whipkey RR, Paris PM, Stewart RD. Drug use in pregnancy. Ann Emerg Med 13(5):346-354 (1984).
171. Levin ED, Uemura E, Bowman RE. Neurobehavioral toxicology of halothane in rats. Neurotoxicol Teratol 13(4):461-470 (1991).
172. Vaughan RW, Sipes IG, Brown B Jr. Role of biotransformation in the toxicity of inhalation anesthetics. Life Sci 23(25):2447-2462 (1978).
173. Berthoud MC, Reilly CS. Adverse effects of general anesthetics. Drug Saf 7(6):434-459 (1992).
174. Wess JA. Reproductive toxicity of ethylene glycol monomethyl ether, ethylene glycol monoethyl ether and their acetates. Scand J Work Environ Health 2:43-45 (1992).
175. Beckman DA, Brent RL. Mechanisms of teratogenesis. Annu Rev Pharmacol Toxicol 24:483-500 (1984).
176. Holmberg PC. Central-nervous-system defects in children born to mothers exposed to organic solvents during pregnancy. Lancet (July 28):177-179 (1979).
177. Eriksson P. DDT and pyrethroids--ecotoxicological considerations. Comp Biochem Physiol C 100(1-2):269-270 (1991).
178. Shuman RM, Leech RW, Alvord E Jr. Neurotoxicity of hexachlorophene in humans. II: A clinicopathological study of 46 premature infants. Arch Neurol 32(5):320-325 (1975).
179. Shuman RM, Leech RW, Alvord E Jr. Neurotoxicity of hexachlorophene in the human. I: A clinicopathologic study of 248 children. Pediatrics 54(6):689-695 (1974).
180. Patterson R, Dykewicz MS, Evans RD, Grammer LC, Greenberger PA, Harris KE, Lawrence ID, Pruzansky JJ, Roberts M, Shaughnessy MA et al. IgG antibody against formaldehyde human serum proteins: a comparison with other IgG antibodies against inhalant proteins and reactive chemicals [Abstract]. J Allergy Clin Immunol 84(3):359-366 (1989).
181. Patterson R, Pateras V, Grammer LC, Harris KE. Human antibodies against formaldehyde-human serum albumin conjugates or human serum albumin in individuals exposed to formaldehyde [Abstract]. Int Arch Allergy Appl Immun 79:53-59 (1986).
182. Broughton A, Thrasher JD, Gard Z. Immunological evaluation of four arc welders exposed to fumes from ignited polyurethane (isocyanate) foam: antibodies and immune profiles. Am J Ind Med 13:463-472 (1988).
183. Israel Y, Macdonald A, Niemelä O, Zamel D, Shami E, Zywulko M, Klajner F, Borgono C. Hypersensitivity to acetaldehyde-protein adducts. Mol Pharmacol 42:711-717 (1992).
184. Kenna JG, Neuberger J, Williams R. Evidence for expression in human liver of halothane-induced neoantigens recognised by antibodies in sera from patients with halothane hepatitis. Hepatology 8:1635-1641 (1988).
185. Israel Y, Niemelä O, Hurwitz E, Arnon R. Monoclonal and polyclonal antibodies against acetaldehyde-containing epitopes in acetaldehyde-protein adducts. Proc Natl Acad Sci USA 83:7923-7927 (1986).
186. Niemela O, Juvonen T, Parkkila S. Immunohistochemical demonstration of acetaldehyde-modified epitopes in human liver after alcohol consumption. J Clin Invest 87:1367-1374 (1991).
187. Niemela O. Acetaldehyde adducts of proteins: diagnostic and pathogenic implications in diseases caused by excessive alcohol consumption. Scand J Clin Lab Invest Suppl 213:45-54 (1993).
188. Niemelä O, Klajner F, Orrego H, Vidins E, Blendis L, Israel Y. Antibodies against acetaldehyde-modified protein epitopes in human alcoholics. Hepatology 7(6):1210-1214 (1987).
189. Niemelä O, Israel Y. Hemoglobin-acetaldehyde adducts in human alcohol abusers. Lab Invest 67(2):246-252 (1992).
190. Prasad AS, Meftah S, Abdallah J, Kaplan J, Brewer GJ, Bach JF, Dardenne M. Serum thymulin in human zinc deficiency. J Clin Invest 82(4):1202-1210 (1988).
191. Coto JA, Hadden EM, Sauro M, Zorn N, Hadden JW. Interleukin 1 regulates secretion of zinc-thymulin by human thymic epithelial cells and its action on T-lymphocyte proliferation and nuclear protein kinase C. Proc Natl Acad Sci USA 89(16):7752-7756 (1992).
192. Taubeneck MW, Daston GP, Rogers JM, Keen CL. Altered maternal zinc metabolism following exposure to diverse developmental toxicants. Reprod Toxicol 8(1):25-40 (1994).
193. Jameson S. Zinc status in pregnancy: the effect of zinc therapy on perinatal mortality, prematurity, and placental ablation. Ann NY Acad Sci 678:178-92 (1993).
194. Kiecolt-Glaser JK, Marucha PT, Malarkey WM, Mercado AM, Glaser R. Slowing of wound healing by psychological stress. Lancet 346:1194-1196 (1995).
195. Sparrow GP. A connective tissue disorder similar to vinyl chloride disease in a patient exposed to perchlorethylene. Clin Exp Dermatol 2:17-22 (1977).
196. Ward AM, Udnoon S, Watkins J, Walker AE, Darke CS. Immunological mechanisms in the pathogenesis of vinyl chloride disease [Abstract]. Br Med J (6015):936-938 (1976).
197. Ward AM. Evidence of an immune complex disorder in vinyl chloride workers. Proc R Soc Med 69(4):289-290 (1976).
198. Griffiths H, Lunec J. Effect of polymorph derived oxidants on IgG in relation to rheumatoid factor binding [Abstract]. Scand J Rheumatol Suppl 75:148-156 (1988).
199. Koshte VL, Aalbers M, Calkhoven PG, Aalberse RC. The potent IgG4-inducing antigen in banana is a mannose-binding lectin, BanLec-I. Int Arch Allergy Immunol 97(1):17-24 (1992).
200. Pusztai A. Dietary lectins are metabolic signals for the gut and modulate immune and hormone functions. Eur J Clin Nutr 47(10):691-699 (1993).
201. Thaler DS. The evolution of genetic intelligence [see comments] [Comments]. Science 264(5156):224-225 (1994).
202. Cairns J, Overbaugh J, Miller S. The origin of mutants. Nature 335:142 (1988).
203. Vitetta ES, Berton MT, Burger C, Kepron M, Lee WT, Yin X. Memory B and T cells. Annu Rev Immunol 9:193-217 (1991).
204. Haeney MR, Goodwin BJ, Barratt ME, Mike N, Asquith P. Soya protein antibodies in man: their occurrence and possible relevance in coeliac disease. J Clin Pathol 35(3):319-322 (1982).
205. Cunningham-Rundles C. Failure of antigen exclusion. In: Food Allergy and Intolerance (Brostoff J, Challacombe SC, eds). Ballière Tindall, 1987;223-236.
206. McGovern JJ Jr, Lazaroni JA, Hicks MF, Adler JC, Cleary P. Food and chemical sensitivity: clinical and immunologic correlates. Arch Otolaryngol 109(May):292-297 (1983).
207. Castle L, Kelly M, Gilbert J. Migration of mineral hydrocarbons into foods. 2: Polystyrene, ABS, and waxed paperboard containers for dairy products. Food Addit Contam 10(2):167-174 (1993).
208. Page BD, Lacroix GM. The occurrence of phthalate ester and di-2-ethylhexyl adipate plasticizers in Canadian packaging and food sampled in 1985-1989: a survey. Food Addit Contam 12(1):129-151 (1995).
209. Conacher HB, Page BD, Ryan JJ. Industrial chemical contamination of foods. Food Addit Contam 10(1):129-143 (1993).
210. Nerin C, Cacho J, Gancedo P. Plasticizers from printing inks in a selection of food packagings and their migration to food. Food Addit Contam 10(4):453-460 (1993).
211. Forsyth DS, Dabeka R, Sun WF, Dalglish K. Speciation of organotins in poly(vinyl chloride) products. Food Addit Contam 10(5):531-540 (1993).
212. Varner SL, Hollifield HC, Andrzejewski D. Determination of benzene in polypropylene food-packaging materials and food-contact paraffin waxes. J Assoc Off Anal Chem 74(2):367-374 (1991).
213. Grob K, Lanfranchi JE, Artho A. Determination of food contamination by mineral oil from jute sacks using coupled LC-GC. J Assoc Off Anal Chem 74:506-512 (1991).
214. Withey JR, Collins PG. Styrene monomer in foods a limited Canadian survey. Bull Environ Contam Toxicol 19(1):86-94 (1978).
215. Ishiwata H, Inoue T, Yoshihira K. Tetramethylsuccinonitrile in polyvinyl chloride products for food and its release into food-simulating solvents. Z Lebensm Unters Forsch 185(1):39-42 (1987).
216. Ahmed FE, Hattis D, Wolke RE, Steinman D. Risk assessment and management of chemical contaminants in fishery products consumed in the USA [Abstract]. J Appl Toxicol 13(6):395-410 (1993).
217. Barlow SM. The role of the Scientific Committee for Food in evaluating plastics for packaging [Abstract]. Food Addit Contam 11(2):249-259 (1994).
218. Gilbert J, Castle L, Jickells SM, Sharman M. Current research on food contact materials undertaken by the UK Ministry of Agriculture, Fisheries and Food [Abstract]. Food Addit Contam 11(2):231-240 (1994).
219. Ziem GE, Castleman BE. Threshold limit values: historical perspectives and current practice. J Occup Med 31(11):910-918 (1989).
220. Norback D, Torgen M. A longitudinal study relating carpeting with sick building syndrome. Environ Int 15:129-135 (1989).
221. Welch LS, Sokas R. Development of multiple chemical sensitivity after an outbreak of sick-building syndrome. Toxicol Ind Health 8(4):47-50 (1992).
222. Molhave L. Indoor air pollution due to organic gases and vapours of solvents in building materials. Environ Int 8:117-127 (1982).
223. Molhave L, Bach B, Pedersen OF. Dose-response relation of volatile organic compounds in the sick building syndrome. Clin Ecol 4(2):52-56 (1986).
224. Montgomery MR, Reasor MJ. A toxicologic approach for evaluating cases of sick building syndrome or multiple chemical sensitivity [Abstract]. J Allergy Clin Immunol 94(2 Pt 2):371-375 (1994).
225. Ipsen J, Deane M, Igenito FE. Relationships of acute respiratory disease to atmospheric pollution and meteorological conditions. Arch Environ Health 18:462-472 (1969).
226. Ito K, Thurston GD, Hayes C, Lippmann M. Associations of London, England, daily mortality with particulate matter, sulfur dioxide, and acidic aerosol pollution. Arch Environ Health 48(4):213-220 (1993).
227. Oppelt ET. Air emissions from the incineration of hazardous waste. Toxicol Ind Health 6(5):23-51 (1990).
228. Pearlman ME, Finklea JF, Creason JP, Shy CM, Young MM, Horton JM. Nitrogen dioxide and lower respiratory illness. Pediatrics 47(2):391-398 (1971).
229. Seaton A, MacNee W, Donaldson K, Godden D. Particulate air pollution and acute health effects. Lancet 345:176-178 (1995).
230. Dumont R. Volatile organic compound survey and summarization of results. Saskatoon, Saskatchewan:Saskatchewan Research Council, 1992.
231. Winter R. A Consumer's Dictionary of Food Additives. New York:Crown, 1994.
232. Ashford A, Miller CS. Origins of multiple chemical sensitivity and effects on health. In: Chemical Exposures: Low Levels and High Stakes. New York:Van Nostrand Reinhold, 1991;59-84.
233. Landrigan PJ. Critical assessment of epidemiologic studies on the human carcinogenicity of 1,3-butadiene. Environ Health Perspect 86:143-147 (1990).
234. Arce GT, Vincent DR, Cunningham MJ, Choy WN, Sarrif AM. In vitro and in vivo genotoxicity of 1,3-butadiene and metabolites. Environ Health Perspect 86:75-78 (1990).
235. Melnick RL, Huff J, Bird MG, Acquavella JF. 1,3-Butadiene: toxicity and carcinogenicity in laboratory animals and in humans. Environ Health Perspect 86:3-5 (1990).
236. Morrissey RE, Schwetz P, Sikov MR, Hardin BD, McClanahan BJ, Decker JR, Mast TJ. Overview of reproductive and developmental toxicity studies of 1,3-butandiene in rodents. Environ Health Perspect 86:79-84 (1990).
237. Lieber CS. Biochemical and molecular basis of alcohol-induced injury to liver and other tissues. N Engl J Med 319(25):1639-1650 (1988).
238. Lambert B, He S. DNA and chromosome damage induced by acetaldehyde in human lymphocytes in vitro. Ann NY Acad Sci 534:369 (1988).
239. Levine SA, Reinhardt MS. Biochemical-pathology initiated by free radicals, oxidant chemicals, and therapeutic drugs in the etiology of chemical hypersensitivity disease. J Orthomol Psych 12(3):166-183 (1983).
240. Truss CO. Metabolic abnormalities in patients with chronic candidiasis: the acetaldehyde hypothesis. J Orthomol Psych 13(2):66-93 (1984).
241. Raucy JL, Kraner JC, Lasker JM. Bioactivation of halogenated hydrocarbons by cytochrome P4502E1. Crit Rev Toxicol 23(1):1-20 (1993).
242. LaMarte FP, Merchant JA, Casale TB. Acute systemic reactions to carbonless copy paper associated with histamine release. JAMA 260(2):242-243 (1988).
243. Marks JG Jr, Trautlein JJ, Zwillich CW, Demers LM. Contact urticaria and airway obstruction from carbonless copy paper. JAMA 252(8):1038-1040 (1984).
244. Schwope AD, Till DE, Ehntholt DJ, Sidman KR, Whelan RH, Schwartz PS, Reid RC. Migration of BHT and Irganox 1010 from low-density polyethylene (LDPE) to foods and food-simulating liquids. Food Chem Toxicol 25(4):317-326 (1987).
245. Savolainen H. Some aspects of the mechanisms by which industrial solvents produce neurotoxic effects. Chem Biol Interact 18:1-10 (1977).
246. Grasso P. Neurotoxic and neurobehavioral effects of organic solvents on the nervous system. Occup Med 3(3):525-539 (1988).
247. Eley BM, Cox SW. The release, absorption and possible health effects of mercury from dental amalgam: a review of recent findings. Br Dent J 175(10):355-362 (1993).
248. Swartzendruber DE. The possible relationship between mercury from dental amalgam and diseases. I: Effects within the oral cavity. Med Hypotheses 41(1):31-34 (1993).
249. Hahn LJ, Kloiber R, Vimy MJ, Takahashi Y, Lorscheider FL. Dental silver tooth fillings: a source of mercury exposure revealed by whole-body image scan and tissue analysis [see comments]. FASEB J 3(14):2641-2646 (1989).
250. Hultman P, Johansson U, Turley SJ, Lindh U, Eneström S, Pollard KM. Adverse immunological effects and autoimmunity induced by dental amalgamas and alloy in mice. FASEB J 8:1183-1190 (1994).
251. Enestrom S, Hultman P. Does amalgam affect the immune system? A controversial issue. Int Arch Allergy Immunol 106(3):180-203 (1995).
252. Lorscheider FL, Vimy MJ. Evaluation of the safety issue of mercury release from dental fillings. FASEB J 7(15):1432-1433 (1993).
253. WHO. Inorganic Mercury. Environmental Health Criteria 118. Geneva:World Health Organization, 1991;107.
254. Reinhardt JW. Side-effects: mercury contribution to body burden from dental amalgam. Adv Dent Res 6:110-113 (1992).
255. Weiner JA, Nylander M, Berglund F. Does mercury from amalgam restorations constitute a health hazard? Sci Total Environ 99:1-22 (1990).
256. Ratcliffe HE, Swanson GM. Human exposure to mercury: a critical assessment of the evidence of adverse health effects. J Toxicol Environ Health 49:221-270 (1996).
257. Siblerud RL. The relationship between mercury from dental amalgam and oral cavity health [see comments]. Ann Dent 49(2):6-10 (1990).
258. Summers AO, Wireman J, Vimy MJ, Lorscheider FL, Marshall B, Levy SB, Bennett S, Billard L. Mercury released from dental silver fillings provokes an increase in mercury- and antibiotic-resistant bacteria in oral and intestinal floras of primates [see comments]. Antimicrob Agents Chemother 37(4):825-834 (1993).
259. Siblerud RL, Kienholz E. Evidence that mercury from silver dental fillings may be an etiological factor in multiple sclerosis [Abstract]. Sci Total Environ 142(3):191-205 (1994).
260. Siblerud RL. A comparison of mental health of multiple sclerosis patients with silver/mercury dental fillings and those with fillings removed [Abstract]. Psychol Rep 70(3 Pt 2):1139-1151 (1992).
261. Siblerud RL, Motl J, Kienholz E. Psychometric evidence that mercury from silver dental fillings may be an etiological factor in depression, excessive anger, and anxiety [Abstract]. Psychol Rep 74(1):67-80 (1994).
262. Siblerud RL. The relationship between mercury from dental amalgam and the cardiovascular system. Sci Total Environ 99(1-2):23-35 (1990).
263. Fitzgerald WF, Clarkson TW. Mercury and monomethylmercury: present and future concerns. Environ Health Perspect 96:159-166 (1991).
264. Svensson BG, Schutz A, Nilsson A, Akesson I, Akesson B, Skerfving S. Fish as a source of exposure to mercury and selenium. Sci Total Environ 126(1-2):61-74 (1992).
265. Lorscheider FL, Vimy MJ. Mercury exposure from interior latex paint [Letter; comment]. N Engl J Med 324(12):851-852 (1991).
266. Beusterien KM, Etzel RA, Agocs MM, Egeland GM, Socie EM, Rouse MA, Mortensen BK. Indoor air mercury concentrations following application of interior latex paint. Arch Environ Contam Toxicol 21(1):62-64 (1991).
267. Agocs MM, Etzel RA, Parrish RG, Paschal DC, Campagna PR, Cohen DS, Kilbourne EM, Hesse JL. Mercury exposure from interior latex paint [see comments]. N Engl J Med 323(16):1096-1101 (1990).
268. WHO. Methylmercury. Environmental Health Criteria 101. Geneva:World Health Organization, 1990;1-144.
269. Balazs T. Immunogenetically controlled autoimmune reactions induced by mercury, gold and d-penicillamine in laboratory animals: a review from the vantage point of premarketing safety studies [Abstract]. Toxicol Ind Health 3(3):331-336 (1987).
270. Waalkes MP, Coogan TP, Barter RA. Toxicological principles of metal carcinogenesis with special emphasis on cadmium. Crit Rev Toxicol 22(3-4):175-201 (1992).
271. Jones MM, Cherian MG. The search for chelate antagonists for chronic cadmium intoxication. Toxicology 62(1):1-25 (1990).
272. Drasch G. An increase of cadmium body burden for this century-an investigation of human tissues. Sci Total Environ 26:111-119 (1983).
273. Elinder CG. Cadmium concentration in samples of human kidney cortex from the 19th century. Ambio 6(5):270-272 (1977).
274. Ragan HA, Mast TJ. Cadmium inhalation and male reproductive toxicity. Rev Environ Contam Toxicol 114:1-22 (1990).
275. Kuhnert BR, Kuhnert P, Debonne S, Williams TG. The relationship between cadmium, zinc and birth weight in pregnant women who smoke. Am J Obstet Gynecol 157:1247-1251 (1987).
276. Kuhnert BR, Kuhnert PM, Groh-Wargo SL, Webster S, Erhard P, Lazebnik N. Smoking alters the relationship between maternal zinc intake and biochemical indices of fetal zinc status. Am J Clin Nutr 55:981-984 (1992).
277. Baghurst PA, McMichael AJ, Wigg NR, Vimpani GV, Robertson EF, Roberts RJ, Tong SL. Environmental exposure to lead and children's intelligence at the age of seven years. The Port Pirie cohort study [see comments] [Abstract]. N Engl J Med. 327(18):1279-1284 (1992).
278. Constantinidis J. The hypothesis of zinc deficiency in the pathogenesis of neurofibrillary tangles. Med Hypotheses 35(4):319-323 (1991).
279. Marsh DO, Myers GJ, Clarkson TW, Amin-Zaki L, Tikriti S, Majeed MA. Fetal methylmercury poisoning: clinical and toxicological data on 29 cases. Ann Neurol 7(4):348-353 (1980).
280. Dafny N, Lee JR, Dougherty PM. Immune response products alter CNS activity: interferon modulates central opioid functions. J Neurosci Res 19:130-139 (1988).
281. Sternberg EM, Chrousos GP, Wilder RL, Gold PW. The stress response and the regulation of inflammatory disease. Ann Intern Med 117(10):854-866 (1992).
282. Stead RH. Innervation of mucosal immune cells in the gastrointestinal tract. Reg Immunol 4(2):91-99 (1992).
283. Payan DG, Goetzl EJ. Dual roles of substance P: modulator of immune and neuroendocrine functions. Ann NY Acad Sci 512:465-475 (1987).
284. Jonakait GM, Schotland S, Hart RP. Interleukin-1 specifically increases substance P in injured sympathetic ganglia. Ann NY Acad Sci 594:222-230 (1990).
285. Ashford A, Miller CS. Chemical Exposures: Low Levels and High Stakes. New York:Van Nostrand Reinhold, 1991.
286. Staudenmayer H, Selner JC, Buhr MP. Double-blind provocation chamber challenges in 20 patients presenting with multiple chemical sensitivity. Regul Toxicol Pharmacol 18(1):44-53 (1993).
287. Tollefson L. Multiple chemical sensitivity: controlled scientific studies as proof of causation. Regul Toxicol Pharmacol 18(1):32-43 (1993).
288. Dinarello CA. Interleukin-1. Rev Infect Dis 6(1):51-95 (1984).
289. Farrar WL, Kilian PL, Ruff MR, Hill JM, Pert CP. Visualization and characterization of interleukin-1 receptors in brain. J Immunol 139:459-463 (1987).
290. Kenney JS, Baker C, Welch MR, Altman LC. Synthesis of interleukin-1 alpha, interleukin-6, and interleukin-8 by cultured human nasal epithelial cells. J Allergy Clin Immunol 93(6):1060-1067 (1994).
291. Weiss B. Low-level chemical sensitivity: a perspective from behavioral toxicology. Toxicol Ind Health 10(4-5):605-617 (1994).
292. Weiss B. Experimental strategies for research on multiple chemical sensitivity. Environ Health Perspect 105(Suppl 2):487-494 (1997).
293 Cohen N, Kehrl H, Berglund B, O'Leary A, Ross G, Seltzer J, Weisel C. Psychoneuroimmunology. Environ Health Perspect 105(Suppl 2):527-529 (1997).
294. Miller CS. Chemical sensitivity: symptom, syndrome or mechanism for disease? Toxicology 111:69-86 (1996).
295. Siegel S, Kreutzer R. Pavlovian conditioning and multiple chemical sensitivity. Environ Health Perspect 105(Suppl 2):521-526 (1997).
296. Russek LG, Schwartz GE. Narrative descriptions of parental love and caring predict health status in midlife: a 35-year follow-up of the Harvard mastery of stress study. Altern Ther 2(6):55-62 (1996).
297. Sorg BA, Hooks MS, Kalivas PW. Neuroanatomy and neurochemical mechanisms of time-dependent sensitization. Toxicol Ind Health 10(4-5):369-386 (1994).
298. Sorg B, Doty R, Antelman S, Miller C. Questions and answers #2. Toxicol Ind Health 10(4-5):387-389 (1994).
299. Hou S, Hyland L, Ryan KW, Portner A, Doherty PC. Virus-specific CD8+ T-cell memory determined by clonal burst size. Nature 369(23 June):652 (1994).
300. Brayton CF. Dimethyl sulfoxide (DMSO): a review. Cornell Vet 76(1):61-90 (1986).
301. Boosalis MG, Solem LD, McCall JT, Ahrenholz DH, McClain CJ. Serum zinc response in thermal injury. J Am Coll Nutr 7(1):69-76 (1988).
302. Antelman SM. One brief exposure to a psychological stressor induces long-lasting, time-dependent sensitization of both the cateleptic and neurochemical responses to haloperidol. Life Sci 51:261-266 (1992).
303. Dantzer R, Kelley KW. Stress and immunity: an integrated view of relationships between the brain and the immune system. Life Sci 44:1995-2008 (1989).
304. Wilder R. Mechanisms of neuroendocrine effects on inflammation and the immune response, pp 860-861. In: Sternberg EM, Moderator, The Stress Response and the Regulation of Inflammatory Disease. Ann Intern Med 117(10):854-866 (1992).
305. Cocke R, Moynihan JA, Cohen N, Grota LJ, Ader R. Exposure to conspecific alarm chemosignals alters immune responses in BALB/c mice. Brain Behav Immunol 7(1):36-46 (1993).
306. Ottaway CA, Husband AJ. Central nervous system influences on lymphocyte migration. Brain Behav Immunol 6(2):97-116 (1992).
307. Meggs WJ. RADS and RUDS--the toxic induction of asthma and rhinitis. J Toxicol Clin Toxicol 32(5):487-501 (1994).
308. Meggs WJ. Neurogenic inflammation and sensitivity to environmental chemicals. Environ Health Perspect 101(3):234-238 (1993).
309. Rossman TG, Roy NK, Lin WC. Is cadmium genotoxic? In: Cadmium in the Human Environment: Toxicity and Carcinogenicity (Nordberg GF, Herber RFM, Alessio L, eds). IARC Sci Publ 118. Lyon:International Agency for Research on Cancer, 1992;367-375.
310. Cooper P. A whiff of petrol. Food Cosmet Toxicol 18(4):433-434 (1980).
311. Riihimäki V, Savolainen K. Human exposure to m-xylene. Kinetics and acute effects on the central nervous system. Ann Occup Hyg 23:411-422 (1980).
312. Randolph TG, Moss RW. Hidden addictions. In: An Alternative Approach to Allergies. New York:Harper & Row, 1980;19-35.
313. Randolph TG, Moss RW. The problems of chemical susceptibility. In: An Alternative Approach to Allergies. New York:Harper & Row, 1980;49-59.
Last Update: March 18, 1998

