Reviews in
Environmental Health, 1998
Environmental
Health Perspectives 106, Supplement 1, February 1998
Key words: primary cell cultures, cell lines, central nervous system, peripheral nervous system, neuron, astrocyte, glia, oligodendroglia, astrocytomas, glutamate, neuropathy target esterase, organophosphate, reaggregate cell culture, organ culture, tissue slice, glioma cell line, neuroblastoma cell line, PC-12 cells, cytotoxicity, myelin, axonal transport, blood-brain barrier, neurotransmitter, calcium
Manuscript received at EHP 9 June 1997; accepted 21 July 1997.
1National Institute of Environmental Health Sciences, Research Triangle Park, North Carolina; 2Penn State University College of Medicine, Department of Pharmacology, Hershey, Pennsylvania; 3Institute fur Toxikologie, University of Zurich & ETH, Schwerzenbach, Switzerland; 4The Scripps Research Institute, LaJolla, California; 5CIBA-GEIGY Ltd., Toxicology Services, Stein, Switzerland; 6Chemical Industry Institute of Toxicology, Research Triangle Park, North Carolina; 7Universita Degli Studi Di Milano Facolta Di Farmacia, Milano, Italy; 8MRC Toxicology Unit, University of Leicester, Leicester, United Kingdom; 9University of Glasgow, Glasgow, Scotland; 10U.S. Environmental Protection Agency, National Health Environment Effects Research Laboratory, Research Triangle Park, North Carolina
Sponsored by the United Nations Environment Programme, World Health Organization, International Labour Organization, International Programme on Chemical Safety.
Address correspondence to Dr. G.J. Harry, NIEHS, PO Box 12233, Research Triangle Park, NC. Telephone: (919) 541-0927; Fax: (919) 541-7721. E-mail: harry@niehs.nih.gov
Abbreviations used: ACh, acetylcholine; 5-HT, serotonin; AChE, acetylcholinesterase; BBB, blood-brain barrier; CNS, central nervous system; DMSO, dimethyl sulfoxide; E12, embryonic day 12; GABA,-aminobutyric acid; GFAP, glial fibrillary acidic protein; GGTP,
Neurons are highly specialized cells and are responsible for the reception, integration, transmission, and storage of information. Afferent neuronal pathways carry information into the nervous system for processing; efferent pathways carry commands to the periphery. In addition, there are interneurons that process local information and transfer data within the nervous system. Within the CNS, neurons are segregated into functionally related nuclei that form interconnecting bundles of axonal fibers. Higher organizational levels consisting of several functionally related neurons are frequently called systems, e.g., motor, visual, associative, and neuroendocrine systems. The CNS consists of a number of systems responsible for the coordination of receiving and processing information from the environment, maintaining the balance of all other organ systems, and responding to changes in the environment.
The in vivo reactions of neurons to injury vary dramatically. Degeneration can be induced by a direct action on the perikaryon or loss of synaptic target site influences and deprivation of trophic factors. A number of chemicals appear to have distinct cellular specificity for neuronal populations. Although specificity can exist and the pattern of degeneration has been used in diagnostic neuropathology, degeneration of any particular neuronal type cannot necessarily identify the damaging agent. Often this pattern reflects the severity and duration of the injury and the acuteness or chronic nature of exposure. The degenerative process of the nerve cell can be either relatively quick or a slow, prolonged process, depending on the underlying mechanism.
Unlike neurons, glial cells have no true synaptic contacts; however, receptors for several neurotransmitters are present and functional on various glial cells. Cell-cell contacts exist between the glial cells and neurons that may regulate both neuronal and glial differentiation and are critical for the glial-guided migration of neurons during development. Glial cells have a dynamic nature and provide critical processes necessary to maintain normal functioning of the nervous system (e.g., regulation of local pH and ionic balances, and tropic support for neurite extension in the form of growth factors and cell adhesion factors). They can also be the target for a toxic response. For either the oligodendrocyte or the Schwann cell, the myelin membrane is vulnerable to numerous substances, toxic agents, and demyelinating autoimmune diseases such as multiple sclerosis and Guillian-Barre syndrome that specifically target and break down the myelin sheath. After injury to the nervous system, the astrocyte and the microglia both display profound responses. The astrocyte responds by proliferation or a morphological change characterized by cellular hypertrophy. This process of reactive gliosis has been proposed as an early marker of damage to the nervous system and can be detected morphologically or quantified by measurements of an astrocyte-specific protein, glial fibrillary acidic protein (GFAP). The sequence of the response can be rapid and transient or progressive and sustained over an extended period of time. The microglial cell assumes an immunologic role and is seen in the normal brain in a resting state; however, with injury it can become mobile and assume macrophage-like characteristics. In the PNS, this phagocytic role is provided by the resident macrophage as well as by the infiltrating peripheral macrophages. In a toxicant-induced response of the nervous system, each cell type may contribute to the overall manifestation of neurotoxicity not only individually but based upon complex cell-cell interactions altered or initiated during the injury response.
Various types of in vitro approaches produce data for evaluating potential and known neurotoxic substances, including primary cell cultures, cell lines, and cloned cells. Although such procedures are important in studying the mechanism of action of toxic agents, their use in hazard identification in human health risk assessment has not been widely accepted. For example, the proposed guidelines for neurotoxicity risk assessment published by the the U.S. Environmental Protection Agency (U.S. EPA) (11) raise concerns about the ability of in vitro techniques to predict the neurotoxicity of various agents found in humans and animal models. This validation step requires considerations in study design, including defined end points of toxicity and an understanding of how a test chemical would be handled in vitro as compared to the intact organism. The neurotoxicity risk assessment guidelines propose that demonstrated neurotoxicity in vitro in the absence of in vivo data is suggestive, but inadequate evidence of a neurotoxic effect. Thus, the in vitro data is used often to enhance the reliability of in vivo data.
Although the assessment of neurotoxic end points in the whole animal are presumed to be causally related to those initiated at the cellular level, in many cases the cascade of effects is not well understood. Chemicals rarely if ever affect all neurons indiscriminately but instead induce selective damage. Cellular toxicity depends on the specific sensitivity of the cell as well as on the extracellular concentration of the test compound. The effects seen may not be due to the primary toxicant administered but rather the result of bioactivation with subsequent response to a metabolite. In addition, toxic responses of neurons in vivo may be the result of toxicity of nonneuronal or nonneural cells, e.g., glial cells, endothelial cells. Observed responses may even be due to a systemic biological process activated by exposure such as hepatic encephalopathy or may be observed only with maturation or aging of the system. These features create a major problem in selecting an in vitro system that will model the nervous system target site or process for any particular chemical. With the use of in vitro systems, it must be kept in mind that no existing in vitro system will be able to measure all chemically induced effects. This limitation is due partly to the lack of all possible target sites and the synergistic interaction between the cell types. In addition, the limited culture life may not allow for the appearance of delayed responses or allow for discrimination between transient and persistent effects.
Many chemicals are introduced for industrial use with limited knowledge of how they might affect a biological organism. Alteration in structure of industrial or agriculture chemicals may appear trivial from a chemical perspective, e.g., addition or substitution of a methyl group, but that same change may have dramatic consequences with regard to activity in biological systems. In general, safety testing is designed to reduce unnecessary exposure of human and other animal populations. Thus, the optimal system for hazard identification would be the one most closely resembling the phenotype of concern in biochemical, physiological, molecular, and other cellular and organ processes. Doseresponse data obtained from in vitro studies must be used with caution because of significant differences in the pharmacokinetic behavior of chemicals observed in vivo and in vitro. In addition, effects observed in vitro are not necessarily predictive of whether an adverse effect will occur following exposure in vivo. However, mechanism(s) of neurotoxicity identified using in vitro studies may contribute to determining whether an effect observed in vitro may also result in an adverse effect in animals or humans following chemical exposure.

Access to the Cellular Environment. The physicochemical environment of cells is easily manipulated in vitro. Substances can be added or withdrawn from the culture medium, allowing precise temporal analysis of the sequence of events. The concentration of the test chemical can be controlled in terms of the amount being delivered to the entire cell population or to an individual target cell; however, this concentration must be consistent with the in vivo level of exposure to be meaningful. In addition, the compound should be presented to the cells in a physiological formulation. Techniques exist that allow the direct injection of a substance into a cell and examination of the resulting cellular responses. However, the use of such techniques is limited to specific experimental questions, given the nonphysiological administration, the possible localized distribution of the chemical, and the injury induced by the injection alone.
Physicochemical properties of chemicals, such as solubility, volatility, pH, binding to components of the culture medium including protein binding, and osmolarity, must be considered prior to use in culture systems. It is difficult to evaluate in vitro chemicals that are either insoluble in aqueous systems or at neutral pH, thus forming insoluble particulate or precipitate over time. To expose cells to such insoluble compounds, additives like dimethyl sulfoxide (DMSO), Tween-80, or ethanol, are often added to the culture medium and could distort the toxicological characteristics. Some investigators have attempted to overcome these problems by solublizing the compounds using physiological carriers such as albumin, lipoproteins, or a specific carrier. Many other chemicals may be volatile and evaporate quickly. Such physiochemical properties present major problems in terms of obtaining adequate and constant concentrations of the compound of interest. The chemical nature of the substance administered must be considered. Alterations in the osmolarity or pH shifts in the culture medium can occur that are toxic to the cells or could stimulate the precipitation of nutrients from the culture media. The addition of a test chemical to the nutrient fluid could result in a direct reaction between the test compound and a component of the culture medium (e.g., protein denaturation, precipitation), possibly affecting the availability of the toxicant or essential nutrients, and modifying signals to the cultured cells. Any protein-binding properties of test chemicals could directly alter the microenvironment of cultured cells in a fashion that may not occur in vivo.
Chemical Disposition and Metabolism. Toxicokinetics consists of absorption, distribution, metabolism, and excretion of chemicals and their metabolites following exposure. The dose needed to induce toxic effects depends on these pharmacokinetic parameters. For example, the route by which a chemical enters the body can substantially alter the quantity absorbed and consequently modify the dose required to cause toxicity. Metabolism at the site of absorption (e.g., gastrointestinal tract, skin, lung) may determine the quantity of the compound reaching the circulatory system and the type and concentration of possible toxic metabolites. The absorbed compound may be distributed throughout the body by the circulatory system. Within the organism, the compound may be bioactivated and/or detoxified before it is released back into the circulation. Such metabolism and biotransformation of foreign compounds in the body can be affected by various enzymatic processes. The specificity and velocity of each are strongly species dependent (19). The rate of removal of a compound depends not only on metabolism but also on the excretion rate; large species differences in steady-state levels can occur due to differences in elimination rate. Differences in drug metabolism and excretion are widely considered to be an important reason for the large species differences observed in both dose and type of toxic response following drug or chemical exposure.
In culture, a test compound either remains unaltered or is relatively slowly modified, and one can examine the intrinsic toxicity of a substance to a cell in the absence of any metabolites. Many compounds require metabolic activation; others are detoxified by metabolism. For example, methyl n-butyl ketone is metabolized to the active compounds 2,5-hexanedione and 2,5-hexanediol. Xenobiotic inactivation systems are expressed in neural tissue, predominantly in nonneuronal cells (20). In addition, blood enzymes such as organophosphate hydrolyzing plasma esterase can be introduced via serum supplementation of growth media. The combined effect of these modifications can lead to unanticipated reductions in the actual concentration of the test agent. It is critical to understand the in vivo metabolism, distribution, and effects of potential metabolites in order to interpret data obtained from both in vivo studies and tissue culture systems. If the neurotoxic potential of a chemical in vivo is dependent upon metabolic parameters such as biotransformation, lipid-aqueous partitioning, and distribution, then the expression of toxicity in a culture system may be meaningless when related to whole-animal studies.
METABOLIC ACTIVATION SYSTEM. For certain classes of compounds (e.g., cyclophosphamide activated by cytochrome P450-dependent monooxygenase enzymes), the inclusion in the cell cultures of a metabolic-activating system is required to obtain toxic responses. Any artificial system used to simulate normal metabolism of chemicals should preferably be comparable to the in vivo condition. Usually, biochemical activation and detoxification of test substances are simulated in vitro by the addition of enzyme preparations, microsomal additives (e.g., embryo extract or S9 liver fractions), or metabolically active cells; however, the biotransformation processes may be incomplete or proceed by pathways different from those operating in intact organisms. Inclusion of a metabolic activation system, whether a liver microsomal fraction, embryonic microsomes, or isolated cells, requires a consistent source of extract to ensure reproducibility of experimental data. The metabolic pattern is determined by the type of metabolic system and the species and sex from which the metabolic competent cells are isolated. However, it can never take into account the polymorphisms seen for a variety of enzymes, some of which may be relevant to neurotoxicity (e.g., cytochrome P450s or monoamine oxidase [MAO-B]). It also requires the use of a number of animals to generate a bulk preparation for continued use. Results from studies using metabolic activation system must be interpreted in light of known cytotoxicity resulting from the addition of hepatic S9 fractions to neural cultures (21). In many cases metabolism is not limited to the liver, but also can include metabolism by resident cells in the organ tissue of interest. For example, in the nervous system, there is evidence that glial cells are necessary to produce the neurotoxic metabolite N-methyl-4-phenylpyridinium ion (MPP+) from the parent compound 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) (22), to convert glutamate to glutamine (23), and to sequester heavy metals from the neural environment (24). Thus, in vitro models in which the influences of extraneous chemical substances are studied are often deficient in competent metabolic systems operating in the whole organism. Although cell culture allows a precise determination of the toxic exposure delivered to cells, it also poses a difficulty in defining a toxicologically relevant dose.
EXPOSURE DURATION. Chemicals can have various toxic effects on the whole organism, depending on the exposure dose and duration. Toxic effects may be seen shortly after the initiation of exposure (acute effects) or only after some delay. In some cases, signs of neurotoxicity may be observed only after repeated or prolonged exposure. The appearance of such effects may not only depend on the type of chemical and dose used, but may also depend upon the biological processes underlying the toxic response. Certain compounds interact directly with accessible cell components such as the cell membrane or vital enzymes. As a result, these structures will be functionally affected shortly after exposure. For these effects, the peak concentration and exposure schedule are of utmost importance. A delay in the appearance of effects is due to the initiation of an irreversible cascade of reactions, after which cell damage is manifested. Each step within the cascade, however, needs time to initiate the subsequent step, resulting in a delay in the appearance of the toxic response. Progressive effects are induced by the initiation of a cascade of reactions similar to delayed effects; however, the first changes can be observed only after a significant latency from which the damage is progressive intensified. In other cases, a toxic effect may require an accumulation of a chemical within the system for an extended period of time rather than a peak serum concentration. An example is bismuth, which shows no acute effects either in vivo or in vitro even at extreme doses/concentrations. Adverse effects become manifest only after a prolonged exposure period (25).
Depending on the type of in vitro system, neural cells may survive only a few hours or several months; however, each cell type can mature during this period. As a consequence, the exposure window differs among the different culture systems because of differences in survival. The majority of culture systems employed are applicable for examining acute responses but are not readily conducive to examining any effects that are progressive or delayed in nature.
Lack of Homeostatic Mechanisms. Using in vitro systems provides the researcher with the ability to study a discrete nervous system area isolated from normal in vivo homeostatic mechanisms. The use of in vitro techniques allows the exact and highly specific measurement of many fundamental biological processes isolated from other areas and from complicating factors contributed by the whole organism. In vitro systems offer much in the way of assessing mechanistic questions in a defined controlled system; however, it is an artificial system, and the effects seen in vitro must be confirmed or substantiated in vivo. The lack of homeostatic influences, such as the regulatory control of the neuroendocrine system, nutritional support provided by the blood circulation, interactions with adjacent cells, and the intercellular components that create a unique microenvironment, may limit the extrapolation of in vitro results to in vivo situations.
In the whole animal, chemical disposition is influenced by several factors. The presence of an intact blood-brain barrier is a major factor in determining a chemical's access to the brain. The absence of this barrier in culture conditions can be viewed in two ways. It may be advantageous in that the culture condition permits direct contact of a chemical with the cell, allowing assessment of the intrinsic toxicity of the chemical. The lack of a blood-brain barrier in culture, however, can also limit the extrapolation of in vitro results to the in vivo condition. Although in vitro conditions allow a chemical direct contact with nervous system cells, the lack of the in vivo filtering processes associated with the blood-brain barrier or lipid-aqueous partitioning (26) contributes to the artificial nature of the culture exposure. Therefore, although cell culture allows a precise determination of the chemical delivered to cells, it also poses a difficulty in defining toxicologically relevant doses. It must be noted that the blood-brain barrier is incomplete during prenatal stages of development and in specific regions of the nervous system. In addition, the permeability of the blood-brain or blood-nerve barrier may be compromised as a result of chemical exposure to either the test compound or by induction of other modulating factors. This would allow passage of components normally excluded from the nervous system.
Multiple Species Comparisons. In vitro systems offer the opportunity for the investigator to examine similar cell types from multiple species. One can measure responses in cells from various species easily when cell lines are used. The response of human cells can be compared with that of other species to address questions of interspecies selective toxicity. However, comparisons may be limited by the inherent differences in the origin of the cell line that are totally unrelated to species origin. Primary cell cultures are not readily available from human tissue. Human lines are derived from a limited number of progenitor cells. However, the development of new cell lines offers a wider variety of human cells to examine. For example a self-replicating culture HCN-1Aa was recently developed from human cerebral cortical neurons obtained from a patient with unilateral megalencephaly, a developmental neuropathology in which immature neurons continue to proliferate (27).
Heterogeneity of the Nervous System. In vivo, the development and differentiation of the nervous system depend on the interaction between neuronal and non-neuronal cell types. In a neurotoxic response, not all compounds directly interact with individual cells but may cause damage by indirect effects such as modifications of cell-cell interactions. As mentioned earlier, what has been perceived as a major advantage of primary cell cultures is the ability to obtain a culture enriched in one particular cell type. Such an isolated condition is an alien state for neural cells. Much work has focused on obtaining enriched cultures representative of neuronal populations in the brain, spinal cord, and ganglion. With these efforts it has been demonstrated that rarely does a neuronal culture exist in the absence of some glial cells and that dynamic interactions exist between the cell types in culture (28,29). In fact, in many cases neurons survive poorly in the absence of astroglial cells (30). When early postnatal cerebellar astroglial cells are cultured in the absence of neurons, they show a flat, undifferentiated morphology and proliferate rapidly. The addition of neurons to such cultures rapidly arrests glial cell growth and induce glial morphological differentiation into profiles resembling cerebellar astroglia seen in vivo (31). Similar effects are seen in cells isolated from other regions of the brain. Neuronal inhibition of glial cell division requires a neuron:glial cell ratio of at least 4 to 1. If the system falls below that ratio, many of the glial cells remain free of neurons and continue to proliferate, suggesting the need for membrane-membrane interactions. It has been reported that in culture the inhibition of glial cell proliferation by neurons can be modulated by the availability and type of serum in the culture media (32). Similar effects are seen with various astrocytoma cell lines (33). Astrocytomas pare among the most devastating CNS tumors, ranging from the slowly growing astrocytomas to the rapidly growing glioma multiformae. Cell lines from these tumors include rat C6, mouse G26-24, and human U-251, HTB-16, and A-172 cells, among others. Neurons plated on these astrocytoma cells rapidly bind to the astrocytoma cell and inhibit glial cell growth. A dynamic interaction between cell types has been reported with inhibition of glia-induced endothelial differentiation by lead exposure in co-cultures of C6 glioma cells and retinal endothelial cells (34).
An example of cellular interdependency and influence on toxicity is the recent finding of the in vitro toxicity of trimethyltin (TMT), a chemical that produces specific neuronal necrosis in the hippocampus and astrocyte hypertrophy, and increased microglia staining. In a simple comparison of cell cytotoxicity, one finds distinct differences in dose levels required to produce a 50% increase in measurements of membrane permeability. Richter-Landsberg and Besser (35) studied the toxic effects of TMT on primary astrocytes isolated from 1- to 3-day-old neonatal rats and used neutral red assay to determine cell viability. They reported a dose of 2.5 µM TMT for half-maximal cytotoxicity in primary astrocytes treated for 24 hr. The toxicity of TMT on primary cultures derived from 1-day-old rat pups containing both astrocytes and neurons was higher with a half-maximal cytotoxicity in primary glial cells of 5 µM as measured by ethidium-calcein viability assay and by immunofluorescence (36). A third study examining the toxicity of TMT on a mixed glial population in culture (37) reported approximately a 30% decrease in cell viability at 10 µM TMT within 24 hr as measured by neutral red assay and lactate dehydrogenase (LDH) release. Given the steep nature of the dose-response curve for lethality in vivo, these differences in vitro raise concern. Although each study derived cells from rat pups within the designated time of postnatal day 0 to 2, there were several differences in the conditions of the culture. In the Richter-Landsberg and Besser study (35), the standard protocol for isolation of rat brain glial (38) was followed, which required a defined region of the cortex to be used for isolation. The initial cell preparation was grown for 2 weeks in flasks in serum-containing medium, followed by shaking to remove both microglia and oligodendroglia. The remaining cells were replated in 24-well culture dishes and used for toxicity studies within 2 days. In the study by Maier et al. (37), mixed glial prepared from postnatal day 2 rat pups by the method of McCarthy and de Vellis (38) were allowed to grow in tissue culture flasks for 2 weeks in medium containing fetal calf serum. They were then replated into 24-well culture dishes in which they were allowed to mature until 14 days in vitro. This 14- to 21-day period following replating is consistent with all neurobiological studies conducted on astrocytes and can be critical to the maturation state of the cells and the proportion of cells differentiated into oligodendroglia, astrocytes, and microglia. Thus, the difference in sensitivity could be due to the cellular population within each culture and the interactions between each cell type, the brain region from which the cells were derived, the maturation state of the cells at the time of toxicant exposure, the substratum on which the cells were plated, and the presence of serum.
During development, neurons and glial cells may interact by influencing differentiation. The anatomical organization of astrocytes and their ability to take up and metabolize synaptic transmitters suggest multiple functions for astrocytes in synaptic transmission (39-41). Glial cells extraneuronally regulate neurotransmitter concentration, thus potentially influencing the neurons (42-44). Numerous studies on the glutamine-glutamate neuronal-glial cycle suggest that glial cells act as modulators of neuronal activity (45). Glial cells may play an important role in maintenance of neuronal glutamate metabolism in cells that use glutamate as a neurotransmitter. Based upon the cellular localization of glutamine synthetase in glial cells (46,47) and glutaminase in neurons, a cycle has been proposed in which glutamate released by the neuron is taken up into astrocytes by a high-affinity uptake system (48,49). Once in the astrocyte, glutamate is converted to glutamine by glutamine synthetase and released. It is then taken up by the neuron and reconverted by glutaminase into glutamate. Gamma-aminobutyric acid (GABA) is released from astrocytes upon glutamate receptor stimulation (50) and could inhibit neuronal signaling. In models of chemical-induced glutamate excitotoxicity, any examination of neuronal cells in the absence of glia would be problematic. Glial cells contain the enzyme monoamine oxidase B (51-53). Thus, any chemical requiring local metabolism in the brain for its toxicity would not be detected in cultures devoid of glial cells, although such a culture condition is highly unlikely even at a high level of neuronal purity.
The role for astrocytes in neurotoxic responses has been supported in primary cell culture studies. Astroglia accumulate lead from the culture medium and concentrate it to levels as high as 1000-fold above the extracellular level (54). Similar reports exist for sequestration of iron in astroglia (55). Quantification of intracellular metal levels by graphite furnace atomic absorption spectroscopy requires a minimum of 2 million cells per sample, thus limiting the number of samples available for analysis. Of interest are the observations that glia in mixed cultures containing other cell types respond differently to lead than either oligodendroglia or astrocytes in pure cultures (56,57).
Examples in Pharmacology and Toxicology
Practical uses of in vitro systems have been established for development of pharmaceutical agents. In general, pharmacological agents are designed to act by a specific mechanism at a receptor site. Depending on the target site involved, one can develop a specific in vitro system that will allow for testing of drug efficacy and potency. In addition, information acquired during drug design, site of action of the drug being tested, and any confounding sites of action that would change the drug potency could lead to additional testing in focused systems. This is a unique environment in which a drug is designed to work via a specific mechanism; because this information is known, in vitro tests can be designed to evaluate this specific mechanism. The use of in vitro techniques to identify a drug for development, refine a drug's action and detect problems with a drug such that developmental costs can be minimized are all worthwhile in an environment where a mechanism of action, the target cell type(s), and/or cell functions are generally known. However, in vitro tests are not generally amenable for assessing potential unexpected or unknown drug side effects and testing is conducted in animals prior to human clinical trials.
A well-characterized mechanism for neurotoxicity is excess release of the excitatory neurotransmitter glutamate. Glutamate excitotoxicity is mediated, in part, via activation of postsynaptic N-methyl-d-aspartate (NMDA)-type glutamate receptors. Cells that possess these receptors are susceptible to excitotoxicity; excess receptor stimulation leads to prolonged influx of sodium and calcium. This influx triggers a cascade of events that ultimately leads to cell death (58). Several glutamate analogs such as domoic acid also produce an excitotoxic lesion. Domoic acid accumulation in shellfish has caused outbreaks of human amnesia via loss of hippocampal and neocortical neurons, which are sensitive to activation of the glutamate receptor.
Identification and understanding of this mechanism of excitotoxicity has been aided by the use of a variety of in vitro systems. Hippocampal slice preparations and organotypic cultures have been used to study electrophysiologic, biochemical, and pharmacologic consequences of NMDA receptor activation. Primary dispersed hippocampal and neocortical cultures are also used for characterizing excitotoxicants. Finally, both Xenopus oocytes and several neuroblastoma-derived cell lines have been transfected with expression vectors directing production of neurotransmitter receptors. Expression of such receptors in cell lines allows additional characterization of receptor responses in a different cellular context. The key reason for success in studying the NMDA receptor in vitro was the clear identification of the mechanistic target for glutamate agonists and antagonists. Clearly, unknown chemicals can be evaluated for excitotoxic properties in vitro using glutamate receptor-bearing cells. However, other, nonglutamate-mediated toxicity may be missed.
The situation as presented for pharmaceutical agents does not always exist in the development of chemicals used in agriculture or industry. One exception to this statement is certain insecticides designed to act at ion channels in the nerve membrane. The pyrethroid insecticides provide an example of pesticides which are neurotoxic but not cytotoxic. There is a reasonable correlation between increased transmembrane sodium flux and neurotoxicity, but pyrethroids have little or no effect on neuronal viability (59). Another successful application of an in vitro or ex vivo test is the prediction of the delayed neurotoxic potential of certain organophosphorous pesticides and industrial chemicals by measurement of neuropathy target esterase (NTE). The ratio of NTE aging to acetylcholinesterase (AChE) inhibition can be measured in normal brain homogenates or in cell cultures. This provides a prediction of the relative acute toxic and delayed neurotoxic potential of these agents. An illustration of the importance of bioactivation is provided by the organophosphates. The sulfur form (e.g., parathion) has very little toxic potential until bioactivated to the oxon form (e.g., paraoxon). This capacity of bioactivation is lacking in the absence of in vivo mechanisms and therefore remains undetected in a metabolically incompetent ex vivo or in vitro system.
Cellular Heterogeneity. In primary cultures the initial cell population is extremely heterogeneous, and it is impossible to obtain a group of cells in culture that is completely representative of those in vivo. For some specific questions, it may be desirable to have cultures of only one cell type. These cultures can be obtained from distinct brain regions by selection of donor optimal age. Once growing in culture, a cell type of interest can be selected by a variety of methods. For example, a cytotoxic antiserum may be used to kill one class of cells; deletion of a critical hormone could selectively eliminate a cell type. Glia and fibroblasts are frequently decreased by the use of DNA synthesis inhibitors that kill only dividing cells. Other methods, such as those used for glial cells, include manipulation of serum concentration in the media, and differential cell adhesion. All of these methods will aid in the purification process; however, no method has been found to ensure the absolute cell purity of the culture. The ability to provide a cell type-enriched culture offers a primary advantage over dissociated cell cultures. This offers the possibility to make biochemical measurements on large numbers of cells rather than on a single cell. Whether the results obtained from these pure cell cultures will be reflective of the in vivo condition is uncertain.
As growth conditions often may select for some phenotypes versus others,
it is essential that the conditions of dissociation
and culturing be identical for each experiment to ensure reproducibility.
With heterogeneous CNS tissue, it is unlikely that the final cell population
will reflect the initial population. The composition of a culture can change
drastically with time if some of the cells in the population continue to
divide. The more rapidly dividing cells will have a selective advantage
over the nondividing cells. If one is using cloned cells from an original
culture, the clones are limited in the number of times they can divide.
Thus it is necessary to periodically isolate new clones. Phenotypic drift
often occurs and mutant cells are difficult to identify in primary cultures.
Serum versus Chemically Defined Medium. Currently, most cultured cells are kept in media containing serum plus a basic nutrient mixture of required sugars, salts, amino acids, and vitamins. Serum contains additional growth hormones and growth and attachment factors. Attempts to fully mimic these components have been difficult. Chemically defined serum-free media have been devised for many cell lines and primary cell cultures. These media may contain salts, vitamins, amino acids, growth factors, hormones, and adhesion molecules specific to each type of cultured cell, and do not necessarily generalize to other cells (60). Chemically defined media conditions have tempered the troublesome, uncontrollable, and undefined nature of sera, as well as problems of standardization and reproducibility of results with various batches of sera. However, it can be assumed that any extraneous supplementation will be minimal compared to the contribution of various components available in vivo. Because of various developmental factors present in the nervous system and limited knowledge of extracellular influences, fully defined media is very different from the in vivo physiological milieu of the cell.
Given the variety of media and supplements, one needs to be aware of the extreme variations in cellular metabolism and gene expression that can result under different media conditions. For example, the metabolism of epidermal growth factor (EGF) varies as a function of serum. EGF remains on the membrane surface of cells grown in a defined serum-free medium factor but is internalized in the presence of serum. Synthesis of glial fibrillary acidic protein (GFAP) and microtubule-associated protein by neural cells is influenced by both serum concentration and animal source. Adrenal cortical cells grown in medium supplemented with horse serum secrete large amounts of corticosterone but display a different morphology in fetal calf serum and produce only 1% of the steroid as made by cells in horse serum. The phenotype cannot be reversed by manipulation of serum type in the culture medium.
Cell Substratum. The substratum on which cells are cultivated contributes greatly to their metabolism and division. Frequently collagen, fibronectin, poly-D-lysine, polyornithine, and highly sulfonated tissue culture plastic are used as cell substrata. Some cells are anchorage dependent (i.e., require an adhesive surface on which to adhere) whereas others proliferate/differentiate either loosely attached, or in suspension as reaggregates. If cells are grown under conditions in which their state of adhesion is experimentally varied, many biochemical characteristics of the cells also change. For example, poly-D-lysine is used often in neural cell cultures, to increase cell-substratum adhesion. Many times poly-D-lysine is used to assure the continued attachment of cells throughout the washes required in various experiments such as precursor uptake studies, enzyme assays, or binding of isotopically labeled ligands to cell-surface receptors. Even for cell viability studies of dye uptake, maximizing the attachment of viable cells to the substratum is critical to experimental outcome. Although poly-D-lysine is often used, it can cause a number of phenotypic changes in cells, including changes in morphology, ultrastructure, and metabolism. Because cell-poly-D-lysine interaction can alter cAMP synthesis and interfere with normal ion channel functioning, this substratum should not be used with cells for electrophysiological experimentation. These effects are likely due to a direct and global action of the polycation on the cell membrane rather than to any specific modifications of the particular phenotypic trait being examined.
With cells that can be grown either in suspension or attached to a surface, differences in metabolism can be observed. In fibroblasts, various amino acid transport systems are used, depending on whether the cells are attached or suspended. Fibroblasts that are grown attached to a surface contain 5 times as much of the protein tubulin as cells grown in suspension. Substrate-attached fibroblasts are also more sensitive to cytolysis by antisera against the cell surface, although the actual number of cell-surface antigens is the same in both culture conditions. The substratum can regulate cell morphology and division rate. Fibroblast-like cells grown on culture plastic are flat and elongated and divide rapidly. The same cells grown on hydrophobic bacteriological plastic do not attach, are round in shape, and divide very slowly if at all. Experiments conducted on this regulation of division and morphology demonstrated that differences in the cells were not minor; fibroblast-like cells can initiate DNA synthesis only when they are sufficiently flattened on the substratum.
Cell Feeding. During the routine replacement of culture medium or "feeding," problems can arise from inconsistent cell culture techniques. Cellular phenotype can be altered in some cells by the addition of new culture medium or the manipulation associated with medium replacement. Therefore, care must be taken to insure that experimental effects can be distinguished from those caused by the addition of new medium. For example, with the addition of new medium, endothelial cells decrease plasminogen activator activity. This appears to be due to an inhibitory factor in the serum of the medium. It has been proposed that this effect can be circumvented by the use of chemically defined, serum-free culture media; however, with neural cells, addition of new growth factors in added medium or dilution of a released growth compound by media exchange may also present a transient change in the cell phenotype. As a result of culture medium replacement, the pH of the medium may rise transiently above 7.4. The processes induced at this pH are somewhat similar to those induced by growth factors. Partial replacement or methods to limit the need for medium replacement can partially overcome these problems.
Oxygen Tension. Atmospheric oxygen tension is a critical component that must be maintained for the culturing of nerve cells. In the majority of published protocols, the atmosphere of the cell within an incubator is maintained at a ratio of 95% air to 5% carbon dioxide. The viability of cerebral cortical neurons and the proliferation and maturation of astroglia cells is maximized at 10% partial pressure of oxygen (61).
Cell-Cell Interactions. Because of differences in cell-cell interactions, differences in metabolism and degree of differentiation can be observed between cells grown as reaggregates or attached to a surface. A simple example is the dramatic alteration in cellular physiology depending on the aggregation state of cells. An example of cell-cell contact in the regulation of hormone receptor function has been demonstrated in neural retina cells maintained in reaggregate cultures. The cytoplasmic cortisol receptor was decreased and the enzyme glutamine synthetase was not inducible by cortisol. However, in reaggregate cells the cortisol receptors are detectable and the glutamine synthetase is inducible at levels similar to in vivo.
Cell Cycle. The overall rate of protein synthesis is similar between dividing and stationary-phase cells; however, the rate of synthesis for about 40% of specific proteins varies as a function of cell phase. Therefore, experimental protocols involving reagents which alter cell cycle (e.g., antimitotic drugs) must be designed to distinguish between the primary experimental effect and the secondary consequences of altering the growth cycles of the cultures. Cell phenotypic changes are also associated with the cell cycle. Cells vary not only among different growth phases of the culture but also throughout the cell cycle. Under normal proliferative conditions, macromolecular synthesis in exponentially dividing cells is always quantitatively different from that in stationary-phase cells.
Age. The donor animal and duration of the culture period are both critical parameters in many organotypic, reaggregate, and dissociated primary cultures. Most attempts at primary culture of nervous tissue have involved the use of fetal or neonatal material because of the difficulties in initiating cultures from the adult vertebrate CNS. These difficulties arise from the destruction of the neurites during cell isolation/preparation procedures and removal of cells from the projection areas. Because of differences in timing of development in various brain regions, neuronal cultures from each brain region have been successful only within a distinct identified age period. Even within the same brain region, individual cell types require distinct donor ages. For example, from rat brain cerebellum, granule cells are isolated from postnatal day 6 to 8 brain, but successful Purkinje cell cultures require isolation from fetal tissue. Glial cell cultures offer a broader window of age from which successful cultures can be obtained. Greater success has been achieved in culturing components of the PNS where primary cultures may be maintained from adult and even aged donors (62,63).
Neuronal cultures differ dramatically in the composition of glial cells, the maturation of the glia, the proportion of glial cell types, and the biochemical responsiveness of the culture. Although glial cultures can be generated from different ages, it is critical to the interpretation of experimental data that the donor age be maintained within a well-characterized biologic and morphologic window. For cortical astrocytes and oligodendroglia, this is between birth and 2 days postnatal. For each of these cell types, the culture age is critically important with regard to the responsiveness of the culture to manipulation. Although it has been claimed that primary nerve cultures can be maintained for several months by using supplemented nutrient media and a substrate that supports adhesion, it is well known that the metabolic properties of the cells will change over time. Factors of donor age, as well as age of the culture, must be addressed whenever primary culture systems are used to determine pharmacological or toxicological responses to various agents.
Slice cultures are prepared from infant animals with the age of the donor is a critical parameter in the degree of organotypic organization achieved in the final preparation. Usually, tissue is obtained from rats within the first week of life. By that time, a fair degree of tissue-specific cytoarchitecture has already been established and the peak of neuronal migration has passed. Within 2 to 3 weeks in culture, an original slice will thin to form a pseudomonolayer of cells and the degree of maturation and differentiation can be determined prior to experimental manipulation. For ex vivo studies, tissue can be obtained from any age animal; however, the maturation of the region of interest prior to sampling and interpretation of data must be considered.
Continuous cell lines are usually derived from tumorigenic tissue and can be obtained from donors of various ages. In culture, these types of cells have a useful life span of approximately 50 divisions characterized by progressively longer time periods between population doubling. Immortalized cell lines are generated from cell lines of limited life span that undergo crisis after which their growth potential changes and the life span becomes unlimited. Clonal primary cultures are derived from the progeny of a single cell, but the cells are limited in the number of divisions they can undergo. Once the cell line phenotype is established, it does not change; however, with increasing passages it slowly drifts with regard to physiological responsiveness.
The sensitivity of any culture to toxicants can be influenced by the age of the culture. Whether it is the influence of passage number in continuous cell lines, maturation and establishment of connections in a reaggregate culture, or the cellular maturation of primary cell cultures, each culture model requires that any experimental manipulation be conducted at an optimal and consistent age in culture. For example, the toxicity of MPTP to dopaminergic neurons is significantly greater in fully developed dopaminergic neurons and toxicity that is dependent upon ion channel activity will require the maturation of this physiological process for detection and examination.
State of Differentiation. Since many of the sources of tissue for primary cultures are from fetal or early postnatal animals, features of adult differentiated neurons or glial cells may not be present. Examples include cerebellar granule cells and dentate gyrus neurons; both cease division postnatally and do not express ion channels until several days in culture. In neuroblastoma and related cell lines, trophic growth factors alter the state of morphologic differentiation and overall gene expression. Well-characterized examples include nerve growth factor (NGF)-induced differentiation of rat pheochromocytoma (PC-12) cells and retinoic acid-induced differentiation of human neuroblastoma cells (SMS-KCNR; SNSY5Y). In both instances, treated cells extend complex neurites, express new neuronal cytoskeletal elements, and show prominent shifts in gene expression and slowed or arrested cell division. The state of differentiation can clearly affect expression of potential targets of neurotoxicity.
Characterization. In specific cases, one requires a relatively pure culture of neuronal or glial cells. To determine the purity of a culture, one must examine the culture not only microscopically for structural features but also by immunocytochemical methods to identify individual cells contributing to the culture. A problem central to all culture systems is cellular impurities and culture heterogeneity. It is, therefore, essential that both the characteristic morphology and antigenic phenotype of the cultured cells be characterized to permit the unique identification of these cell types. These may include either immunocytochemical or enzymatic markers that are associated with the plathora of CNS cells (Table 2).

One possible contaminating cell in any primary cell culture is the fibroblast. Attempts are made to eliminate this cell type from neural cultures by removing all brain meninges prior to dissociation, preplating for early adherence removal, and adding cytotoxic agents for proliferating cells. The case is similar for glial cells as a contaminating factor in neuronal cultures; however, detection of all types of glial cells requires the use of a number of antibody evaluations. One of the major concerns with contributing glial cells is the surface area of the glia relative to neurons. Although one may determine by counting number of cells that glia contribute less than 5% of the culture, each one of these cells has the surface area of hundreds of neurons. With glial cultures, the use of differential adherence or gradient fractionation aids in the generation of enriched cultures of individual glial cells. However, recent evidence suggests that factors previously contributed by astrocytes are the product of contaminating microglia cells within the culture preparation. As little as 1% contribution of microglia can significantly influence the interpretation of data derived from astrocyte cell cultures. Not only must the culture system be characterized in use with regard to contributing cell types, but the maturation state of the specific cell type of interest must be determined. For example, during maturation, glial cells--both astrocytes and oligodendroglia--will progress through distinct periods of surface antigen expression, and the use of the standard antibody staining to GFAP may not detect all contributing astrocytes.
Surveillance. Antibiotics and/or fungicides often are added to a cell culture preparation to prevent contamination. Such additions can reduce cellular activity and modify drug-induced neurotoxic effects. Unlike bacteria and fungi, contamination of mycoplasma usually does not result in turbid growth or macroscopic alterations of the cells. However, mycoplasma contamination may cause adverse effects such as changes in metabolism, growth, viability, DNA, RNA, protein synthesis, morphology, and virus propagation, thus leading to unreliable experimental data. For example, in a mixed glial preparation containing microglia, any contaminating factor will activate the microglia into phagocytic activity. The source of contamination may originate from initial culturing techniques and in the case of a cell line can be propagated among a number of research laboratories. As a quality control measure, all culture systems should be evaluated for the presence of any bacterial, fungal, or mycoplasmic contamination prior to use.
Other Culture Condition Factors. There are a number of other factors unique to culture systems that raise concern for neurotoxicologists. For example, the viability and plating efficiency (number of cells that attach to the substratum with initial plating) of individual cells is greatly influenced by the technical manipulation of the cell source. For primary cultures, the method of dissociation has a major influence. Enzymatically dissociated cells generally survive better than those dissociated by mechanical means. It has been suggested that at early prenatal stages a mechanical disruption of cells produces greater cell survival and enzymatic disruption produces greater survival of cells isolated from late prenatal and postnatal ages. For cell lines, the storage condition of the cells (e.g., frozen in DMSO or glycine, by quick or slow method, in media or buffer) and methods of plating can influence the proportion of cell viability. For each culture system, these factors can influence greatly the ability to compare experimental data across various laboratories using different techniques or different cell sources.
In summary, culture systems offer the potential advantage of examining a direct cellular response to a chemical, determining biological processes at a level that would not be accessible in vivo, and assessing underlying mechanisms associated with cellular development and functioning of the nervous system. Although chemicals can be easily added and withdrawn from the cultures, and their effects directly probed in culture systems, caution should be used when correlating effects occurring in vitro to those observed in the intact animal, where additive interactions are likely to occur. One must remember several concepts: a) A number of different, sometimes competing, processes influence the ability of a toxin to attack and destroy specific cells. Metabolism of the administered agent by a nontarget cell or tissue may be responsible for bioactivation or detoxification of the compound or its metabolite, affecting the vulnerability of the cells to the neurotoxin. b) A cell culture is simpler and much more homogeneous than any tissue, in particular the CNS. Removal of many cell types and barriers can facilitate diffusion or even active transport of the compound or its metabolite, limiting or enhancing toxicity by determining at which sites the toxin can reach sufficiently high concentrations to interfere with vital cellular processes. c) The capacity of the cell to repair or replace damaged organelles or enzymes can also be critical in determining cell survival after toxic insult and may depend on neighboring cells and physical barriers, which would be absent in the culture.
Although culture systems are relatively convenient to use, their ability to predict the neurotoxicity of a chemical in situ or the potency of a series of structurally related analogs depends on whether the culture system expresses the target for a given neurotoxic effect. Any culture system derived from resident cells of the nervous system represents an artificial system with regard to the in situ condition. Therefore, any experimental use of such systems to address specific scientific questions requires that precautions are taken to ensure biological relevance, validity and reliability of the data. Data generated from in vitro studies must be interpreted within the limits and framework of the experimental design. In vitro studies are relevant only if the target site is contained within the culture model. Any extrapolation to the in vivo condition must take into consideration modulating factors that are present in the whole animal, e.g., blood-brain barrier, lipid partitioning, metabolic activation, or detoxification of a chemical.
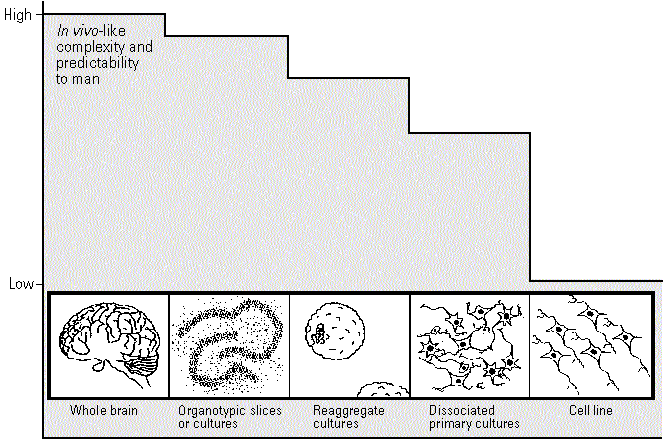
Figure 1. Model culture systems.
Uses and Limitations. For examination of altered nervous system functioning, isolated models have been selected that contain key biochemical and morphological features which are specifically targeted by neurotoxicants in vivo. Each type of tissue culture system to be discussed has its own specific advantages and disadvantages (Table 3). Dissociated cell cultures allow for visualization of individual living cells and for monitoring both morphological and electrophysiological features. Although the histotypic tissue organization is lost as a result of the dissociation procedure, it is believed that neuronal and glial cells migrate to rearrange themselves on the substratum and differentiate according to their function and abilities. However, in vivo-like structures cannot be obtained by this technique. Dissociated cell cultures are more accessible to experimental manipulation than slice cultures and are easier to obtain and maintain. It is possible to obtain and correlate biochemical, morphological, electrophysiological, and molecular data from a single cell. Additional purification methods can be used to enrich a particular cell type in a dissociated cell preparation, but it is difficult to obtain and define pure cultures of any one cell type.

Reaggregate cultures offer a more structured, three-dimensional extracellular space that more closely approximates the in vivo conditions for cell growth and development. Initially, the cells inside an aggregate are distributed in a random fashion followed by reorganization into a pattern formation similar to the in vivo structure. These preparations emphasize the importance of cell contact and histotypic organization by identifying markers induced in specific cells similar to in vivo yet absent in dissociated cell cultures (e.g., glucocorticoid-induced glutamine synthetase in retinal glial Muller cells). In hopes of similar biological advantages, some laboratories use three-dimensional matrix cultures made from components such as collagen and fibronectin. Clonal cell lines of tumoral origin provide homogeneous cell populations in large quantities in a very reproducible manner. Usually the common cell lines of choice are those that continue to express in culture the differentiated properties of their normal cell counterpart.
It must be noted again that these cell culture systems represent cells that are no longer part of an integrated neural network and may develop an altered appearance, metabolism, and an altered response to chemicals. Some believe that these concerns can be addressed by experimental design and choice of end points. No such efforts, however, will compensate for the isolation of a system from the natural neural environment and all of the systemic influences on that environment. The ability of culture systems to predict the neurotoxicity of a chemical in situ or the potency of a series of structurally related analogs can depend on whether they express the target for a neurotoxicant's action.
Types of Culture Systems Used in Neurotoxicology. ORGAN CULTURES. Large parts of the spinal cord, ganglia, or sensory organs (e.g., eye) can be maintained as organ cultures. Other neural organs are difficult to perfuse in a physiological manner, if at all. An example of an organ that has been studied using tissue culture is the eye. In these experiments, the eye is isolated under anesthesia and cannulated at the ophthalmociliary artery (64,65). A test substance can be introduced or washed out by the perfusion solution via the cannula. Effects caused by the administration of a chemical are usually measured by electrophysiological techniques. In the eye the reference electrodes are positioned at the cornea and at the posterior pole. Standing potential, electroretinogram, and light peak recordings are made using a glass pipette placed in the vitreous humor.
It is assumed that the concentration of a test substance at the target cells is comparable with the in vivo situation if the concentration in the perfusate is the same as that in the bloodstream. A further advantage is that electrophysiological effects can be measured without knowing the target cell type. The disadvantage is that for any individual organ, recordings can be made for only a limited period of time. In the case of the eye this time period is a maximum of 6 hr. Therefore, only acute effects can be measured. It is not possible to determine the reversibility of the induced effects beyond this limited time period.
EXPLANT/SLICE CULTURES. Organotypic cultures are derived from explants of relatively undifferentiated embryonic brain, spinal cord, or sensory organs and develop into integrated neuronal and glial populations. For some regions of the nervous system, the age of animals used in explant cultures can vary from embryonic to postnatal periods but must remain constant across comparisons (66). The tissue explant may be a thin slice, a chunk, or the complete organ, as is the case for the sympathetic ganglia. The main advantage of this system is the presence of a three-dimensional organization in which some of the organ's structural and functional characteristics are retained. The organotypic explants/slices can survive in culture for a week to months, depending on the culture conditions. The disadvantage is the difficulty in quantifying small changes in differentiation or viability, depending on the parameters measured. The nutrient supply (e.g., oxygen, media compounds) available to the cells within the slice/explant is dependent on the diffusion rate and is governed by the thickness of the slice/explant. Often, depending on the thickness of the slices after prolonged culture periods, a hypoxia in the center of the tissue resulting in necrosis cannot be precluded. Only in a small layer of the tissue is it possible to maintain in vivo-like physiological conditions.
The goal of this culture technique is to obtain a preparation with a high degree of cellular maturation and differentiation and with an organotypic organization with the ability to assess individual neurons. The roller-tube technique first used with nervous system tissue has allowed preparations to be maintained in vitro for prolonged periods of time. These systems can be used to study processes involved in nervous system development such as specificity of axonal growth and the mechanisms involved in target contact. Organotypic slice cultures retain, at least for a limited period of time, important organizational features of the original tissue and allow for the morphological identification of specific cell types as well as the electrophysiological responses. Given the massive denervation and cell death caused by the explantation procedures and the novel influences of the tissue culture environments, it cannot be assumed that the properties of organotypic cultures are identical to their in vivo counterparts. What can be experimentally exploited, however, is any particular property of the cell that is the same under both in vivo and in vitro conditions. This culture technique has proven worthwhile in the use of sympathetic ganglia cultures to characterize and purify NGF and in the rat hemidiaphragm preparation to study reinnervation and rate of acetylcholine receptor (AChR) turnover. These cultures have been helpful in solving several problems that could not be addressed in animal experiments.
Slice cultures are prepared from infant animals; the age of the donor is a critical parameter with regard to the degree of organotypic organization achieved in the final preparation. Usually, tissue is obtained from rats within the first week of life. Within this time, a fair degree of tissue-specific cytoarchitecture has already been established and the peak of neuronal migration has passed. Within about 2 to 3 weeks in culture, an original 400-µm thick slice will thin to form a pseudomonolayer of cells and with contrast enhancing optic, the individual neurons can be observed in the living state. The results of hypoxia and necrosis can be seen at the center of the explant due to its thickness. Microscopic examination of individual cells is limited by the thickness of the explant. When using slice culture techniques, the degree of maturation and differentiation must be determined under the culture conditions used. There is a high likelihood of synaptic rearrangement which must be examined using histological techniques. Morphological differentiation can be evaluated by cellular injections with dyes such as Lucifer Yellow or horseradish peroxidase.
In many cases, the organotypic culture provides direct access to the neurons and allows placement of stimulating and recording electrodes. A number of features of the slice culture make it attractive for evaluating the electrophysiological activity of individual cells and determining the effects of various pharmacological agents. The presence of functional synapses needs to be assessed by electrophysiological recording techniques that require sophisticated methodologies and critical controls such as temperature. Both the excitatory and the inhibitory systems need to be evaluated. Receptor activity has been studied in various organotypic slice cultures. Such receptor studies include excitatory amino acids, GABA, acetylcholine, norepinephrine, opioid, and growth factor receptors. Pharmacological comparison of these receptors in vivo and in slice preparations show identical characteristics not only in activity but also in cellular distribution. This is not always the case for dissociated neurons.
Although slice cultures display a high degree of neuronal differentiation, this technique does not ensure that all cell types survive or that all important properties such as receptor sensitivity or neuronal connections are established. Although important features of the characteristic cellular and tissue organization of the original tissue are retained, the multitude of connections from various other brain regions is not represented. Loss of afferent input to the slices results in synaptic rearrangement within the slice culture. The absence of both efferent and afferent connections that would normally occur in vivo is a significant factor in determining the types of questions that could be addressed with such a system. This procedure demands from the experimenter an understanding of the morphological characteristics of the region examined.
Explant/slice cultures originate from animals; although a number of slices can be obtained from any one animal, statistical concern requires slices generated from numerous animals. (i.e., animal= statistical unit). Given the various maturation times for each brain region, the optimal age to obtain brain tissue will vary among the different brain regions, limiting the likelihood of collecting various brain regions from any one animal. Additionally, survival time must be considered i.e., the slices must be processed into culture within a defined period of time, usually within 45 min.
Hippocampal Slice Cultures. Preparations are obtained from the brains of 5- to 7-day-old rodents. A 400-micron slice is placed on a specially prepared slide and cultured in roller drums. An estimated number of 10 slices can be obtained from each animal. After several weeks in vitro, the thick slices will flatten to virtual monolayer thickness thus allowing observation of individual neurons under phase contrast microscopy. This flattening is due to a lateral migration of nerve cells that are located in the deeper layered structures. This reorganization is proposed to be due to a limited oxygen supply at the deeper layers. Additionally, any structures adjacent to the cut surface or the severed afferent and efferent fibers degenerate. With the hippocampal slice, the reorganized three-dimensional structure retains the original hippocampal cytoarchitectural relations (67). Morphological differentiation can be maintained similar to in situ conditions for both the pyramidal and granule cells. Synaptic organization can also be maintained; however, aberrant supragranular projections are often reported. Techniques using Golgi impregnation and dye injections allow evaluation of morphological differentiation and dendritic arborization.
In slice culture, neurons and associated cells can be maintained for relatively long periods of time, thus allowing for extending periods of exposure to assess toxicological effects. Some researchers claim that slice cultures can be viable for up to a year; however, the cellular phenotype will change with age. Brain slices, especially the hippocampal slice, have been used extensively for neurobiological studies and the approaches have been adapted for utilization in neurotoxicity evaluation. For example, the hippocampus is vulnerable to excitotoxic agents such as kainic acid, which causes a neuronal necrosis of the pyramidal cells. The excitotoxic effect of kainic acid has also been demonstrated in the hippocampal slice culture, with a selective vulnerability of the CA3 pyramidal cells and no effect on the CA1 neurons, granule cells, and AChE-positive interneurons (68). This ability to detect a selective effect is critical in assessing neurotoxicity because in vivo, rarely are all cells of any one type affected. The basis for neuronal selectivity remains unknown. The hippocampal slice preparation has also been used in assessing the toxic response to bismuth. In agreement with human data, acute exposure had no effect (32). However, chronic application produced morphological evidence of pyramidal cell degeneration. The hippocampal slice preparation has been used to study the effects of organoalkyl metals such as trimethyltin and triethyltin.
In situ, the hippocampus is critically involved in the process of long-term potentiation as a component of synaptic plasticity. This process is of interest in many chemically induced alterations in neural functioning. The hippocampal slice preparation was evaluated electrophysiologically after exposure to the known hippocampal toxicant trimethyltin and showed similar results to in vivo effects (69); however, some important differences were noted. These researchers proposed the use of the hippocampal slice culture preparation as a screen for neurotoxicity which would be able to differentiate effects of chemicals on excitatory and inhibitory systems, mechanisms underlying neuronal plasticity, and regional differences in susceptibility to toxic insult. However, such uses for this test system are well outside the criteria set for a screening test and given the technical demands to confirm the integrity of the slice and the complexity of the hippocampal system, this test system would probably be more useful in addressing specific mechanism-based questions of both basic neurobiological and neurotoxicological nature.
Other Brain Slices. Other brain regions have been cultured successfully by the slice technique including the cerebellum, cortex, and the locus coeruleus. Like the hippocampal slice, the dendritic arborization of neurons derived from the locus coeruleus or cerebral cortex are similar to the in vivo structure. The cultured cerebellar Purkinje cells, however, form multipolar dendrites or dendrites of reduced complexity as compared to the in vivo state. Co-cultured slice cultures have been used to establish an in vitro analog of in vivo axonal connectivity among various brain regions. The ability to determine the success of fiber infiltration and connectivity is dependent upon the selectivity of immunostaining for a particular cell type. For example, in co-cultures of locus coeruleus and cerebellar or hippocampal slices, the invading fibers from the locus coeruleus can be visualized by immunohistochemical staining with antibodies to tyrosine hydroxylase. The co-culture technique has been used to study trophic factor interactions, axonal targeting, and synaptic transmission. It offers a unique method to examine such processes under the influence of various agents; however, the effectiveness of the model will reflect the mechanistic question asked.
PRIMARY CULTURES. Suspension and Reaggregate Cultures. Reaggregate cultures have great utility for examining biochemical end points where a large cell population is needed. Brain tissue is dissociated into a suspension of individual cells and incubated under gyratory conditions to form a confluent monocellular layer with patterns of cell alignment similar to those seen in vivo. Within 1 hr, small cell clumps (reaggregates) are formed. After 2 days these reaggregates have a stable diameter of 0.15 to 0.8 mm depending on the dissociation technique and culture conditions (70-72). Like explants/slices, a general disadvantage of reaggregates is the decreased supply of oxygen and other components of the culture medium to the center of the reaggregate. Dependent on the tissue from which the cells were derived, several layers can be discriminated with reaggregates with patterns of cell alignment similar to those seen in vivo (73-75). In addition, some foci of cells of the same cell type are observed after some days in culture innervating other parts of the reaggregates. The cells undergo morphological differentiation including synaptogenesis and myelination (74,76,77). In a developmentally regulated pattern, the cultures express cellular specific proteins for neurons and glia. Critical to this technique are the age of the donor, the nervous system site extracted, the culture conditions, and the dissociation procedure used. Not only are these features critical to the viability of the culture but also to the degree of cytoarchitectural development. Reaggregate cultures can be maintained by rotation under continuous incubation conditions using chemically defined media for prolonged periods of time (maximum reported >6 months) (72). Thus, the cells can be used immediately in suspension to assess acute biochemical responses or used for future examination of biological responses. Reaggregates have been proposed for use in toxicological studies (78,79).
Dissociated Primary Cultures. The dissociated primary culture is the most widely used in vitro system in neurobiology. Typically, tissue is removed from the embryonic or fetal animal and dissociated mechanically or by proteolytic enzymes. The cells then are washed and placed in culture dishes. Usually, within an hour living cells will adhere to the culture dish substratum. Cells of the CNS are anchorage dependent and those that do not adhere usually die. The fraction of cells which adheres and survives varies greatly with the type of tissue. The plating efficiency is dependent on the dissociation technique, the type of substratum, the culture medium composition, and the type of tissue. A reduced initial survival rate may induce a change in the cell type composition, possibly resulting in an artificial enrichment or depletion of certain cell types. After the initial selection of cells surviving the dissociation process, there is a selection for cells compatible with the culture environment. The presence and amount of serum and trophic factors, oxygen tension, the composition of the substratum, and seeding density strongly affects the viability and differentiation of cultured cells (dedifferentiation, transdifferentiation, differentiation inhibition, induction of differentiation). With CNS tissue, alterations in technical parameters such as age of donor, dissociation techniques, seeding density, and medium composition can greatly influence the characteristics of the culture.
Neuronal primary cultures. Much work has focused on obtaining cultures representative of neuronal populations in the brain, spinal cord, and ganglion. For each of these preparations, the age of the donor is critical for successful culturing of each cell type from different brain regions. Therefore, one is forced to maintain a consistency in donor age in order to generate a culture preparation. The dissection techniques and nutrient media are unique to each level of the neuroaxis and are empirically derived (6,7). For example, preparations of hippocampal neurons are derived from fetal brain tissue where cortical and cerebellar neurons can be derived from postnatal brain tissue. Even within a brain region such as the cerebellum, individual cell types require distinct donor ages. e.g., cerebellar granule cells are isolated from postnatal day 6 to 8 brains whereas successful Purkinje cell cultures require isolation from fetal tissue. Although dissociated cell culture techniques have been successful for numerous brain cell regions and cell types, cultures of dissociated peripheral neurons, sympathetic ganglion cells, and dorsal root ganglion cells have been a more recent addition. This is apparently due to their fastidiousness with respect to their culture media. However, successful conditions have been established and such cultures of the peripheral neurons have been used extensively to study problems of nerve differentiation and electrophysiology. As previously mentioned, whether these cultures will be reflective of the in vivo condition is a question that is continually asked. In many situations this is not the case. For example, Davenport et al. (80) were unable to detect in primary neuronal cultures of either the cortex or the cerebellum the critical in vivo distinction of differential sensitivity to monohalomethanes between cerebellar and cerebral cells.
Glial primary cultures. A number of techniques and media conditions exist for the generation of dissociated primary cell cultures of nervous system glia cells. Usually a mixed glial culture is derived from rat or mouse cortex between birth and postnatal day 2 (38). Between 6 and 18 pups contribute to each culture preparation comprised of three phenotypically distinct glial cells, the astrocyte, the oligodendrocyte, and the microglia. Based upon differential cell adhesion, each cell type can be subcultured to a relatively enriched population. Although much information is available on each individual cell type in culture, only recently have the interactions among the various glial cells been appreciated. In addition, results previously obtained from a pure culture of astrocytes must now be reconsidered in light of new information on the function and responsiveness of other glial cells and techniques used to assess culture purity.
Depending on the age of the donor, cell cultures differ dramatically in the glial cell composition, the maturation process, and biochemical responsiveness. Therefore, it is critical to any interpretation of experimental data that the donor age be maintained within a well-characterized biological and morphological window. For rodent cortical astrocytes and oligodendroglia, this is between birth and day 2 postnatal (38). Individual glial cell cultures can be generated based upon differential attachment of each cell type. Once a mixed glia culture has been established, e.g., in approximately 2 weeks, culture flasks can be shaken (250 rpm) to dislodge both microglia (within 2 hr) and oligodendroglia (18 hr) from the underlying astrocyte monolayer. A microglia culture can be obtained from these mixed glia cultures by plating of the media removed after 2 hr of shaking. Cells are allowed to attach to tissue culture plates over approximately 1 hr. The media is then removed and replaced with fresh media. After shaking, astrocytes can be subcultured by enzymatic detachment, filtration, and an initial plating period of 1 hr to remove any remaining contaminating microglia. Additional steps must be taken to ensure removal of any contaminating microglia from the oligodendroglia subculture. After cellular enrichment, medium conditions are manipulated with 1% fetal calf serum or chemically defined medium to ensure differentiation of progenitor cells to oligodendroglia. The generation of enriched subcultures depends upon a relatively large number of animals.
The majority of experiments using Schwann cell cultures have investigated
the normal interactions between Schwann cell and axons. Three approaches
have been used to isolate Schwann cells for culture. Fresh sciatic nerve
tissue isolated from 2-day-old rat pups undergo enzymatic digestion with
trypsin and collagenase, followed by trituration and plating for 24 hr.
These cultures initially contain both bipolar spindle-shape Schwann cells
and fibroblasts. The rapidly dividing fibroblasts are removed by the addition
of the antimitotic agent cytosine arabinoside for 48 hr followed by removal
for 6 hr and readministration for 24 hr (81). Cells can be be used for
experimentation after being allowed to recover for an additional 24 hr
in normal media. Typically, 0.8 ![]() 106 cells/12 pups can be obtained, with approximately 95% characterized
as Schwann cells. A similar procedure can be used to isolate Schwann cells
from frozen neonatal sciatic nerves (82). Sciatic nerves from 2-day-old
rat pups can be slow frozen in media, DMSO, and fetal calf serum (50%)
and stored in liquid nitrogen. This method differs from the fresh tissue
procedure in that frozen nerve cells require an additional 2 days to recover
from the initial dissociation. Thus, this procedure requires a total of
7 days from dissociation to final use rather than the 5 days required when
using fresh tissue. One major advantage to the use of frozen tissues is
that tissue can be collected over time and stored until a sufficient amount
is obtained for an adequate cultured cell yield. Schwann cells from both
preparations appear identical in morphology and in their response to axonal
membrane mitogens. An immortalized Schwann cell line has been produced
and offers the advantage of rapidly generating extremely large numbers
of Schwann cells that are genetically identical (83). These cells are similar
to primary Schwann cells in their immunoreactivity to Ran-2 and glial fibrillary
acidic protein antibodies and in their ability to segregate individual
neurites in cultured dorsal root ganglia (83). When plated on the artificial
basement membrane matrigel, the cells appear to react similarly to primary
cells; however, their proliferation rate is decreased and the cells take
on a spindle-shape morphology, suggesting a termination of proliferation
and initiation of differentiation (84). Whether primary cells or cell lines,
Schwann cells are very sensitive to the substratum and the serum concentration.
They grow best on laminin or other extracellular matrix substratum and
they require high levels of serum (10%). They do poorly in low serum (1%),
especially in any studies examining a response to chemical exposure.
106 cells/12 pups can be obtained, with approximately 95% characterized
as Schwann cells. A similar procedure can be used to isolate Schwann cells
from frozen neonatal sciatic nerves (82). Sciatic nerves from 2-day-old
rat pups can be slow frozen in media, DMSO, and fetal calf serum (50%)
and stored in liquid nitrogen. This method differs from the fresh tissue
procedure in that frozen nerve cells require an additional 2 days to recover
from the initial dissociation. Thus, this procedure requires a total of
7 days from dissociation to final use rather than the 5 days required when
using fresh tissue. One major advantage to the use of frozen tissues is
that tissue can be collected over time and stored until a sufficient amount
is obtained for an adequate cultured cell yield. Schwann cells from both
preparations appear identical in morphology and in their response to axonal
membrane mitogens. An immortalized Schwann cell line has been produced
and offers the advantage of rapidly generating extremely large numbers
of Schwann cells that are genetically identical (83). These cells are similar
to primary Schwann cells in their immunoreactivity to Ran-2 and glial fibrillary
acidic protein antibodies and in their ability to segregate individual
neurites in cultured dorsal root ganglia (83). When plated on the artificial
basement membrane matrigel, the cells appear to react similarly to primary
cells; however, their proliferation rate is decreased and the cells take
on a spindle-shape morphology, suggesting a termination of proliferation
and initiation of differentiation (84). Whether primary cells or cell lines,
Schwann cells are very sensitive to the substratum and the serum concentration.
They grow best on laminin or other extracellular matrix substratum and
they require high levels of serum (10%). They do poorly in low serum (1%),
especially in any studies examining a response to chemical exposure.
Data suggest that Schwann cell cultures can be used to study the initial proliferative responses and the transduction mechanisms involved in the proliferative response of Schwann cells to axonal membranes (85-90). Schwann cell expression of myelin-related proteins is exquisitely sensitive to axonal contact. In culture Schwann cells do not make a detectable amount of the myelin-specific protein P0 (81). Early experiments examining toxicant-induced alterations in Schwann cell functioning focused on the effects of lead due to the in vivo effect of a demyelinative peripheral polyneuropathy. Pleasure et al. (91) demonstrated a lead-induced decrease in the rate of cell proliferation with no alteration in the ability to differentiate. However, in many cases of peripheral nerve toxicity, the chemical of interest produces a characteristic demyelination in vivo. Although it is possible to establish a co-culture with axons in which Schwann cells produce myelin, these procedures are extremely difficult and the degree of myelination is minimal, unlike the normal in vivo situation. While limited aspects of Schwann cell function can be examined in vitro any interpretation of data within the framework of in vivo toxicant-induced peripheral nerve demyelination is limited if not impossible.
CONTINUOUS CELL LINES. Continuous cell lines are transformed cells derived from neuroblastomas, gliomas, and pheochromocytomas with a useful life span of approximately 50 divisions. Cell lines of limited life span often undergo crisis after which their growth potential changes and their life span becomes unlimited. These cell lines are termed immortalized cell lines. The major attributes of continuous clonal cell lines are homogeneity and the ease with which a large quantity of cells can be grown. Features of established cell lines include the capacity to undergo an unlimited number of cell divisions, altered cell and colony morphology, lack of locomotion, lack of contact inhibition, lack of density-dependent inhibition of cell multiplication, loss of anchorage dependence, and high fibrinolytic activity (92). In addition, once the phenotype of the cell line is established it does not change. However, it has been observed quite often that the cells will drift with regard to physiological responsiveness with increased passages.
Cell lines are available for a number of donor species, but availability of neural cell lines is rather limited. Lines of cells exist that are representative of neurons (neuroblastomas) and glia (oligodendrocytes, Schwannoma, astrocytomas). Because of the numerous limitations of working with tumor-derived cells, cell lines devoid of tumoral properties have been generated with the use of retroviral vectors for transferring oncogenes to glial or neuronal progenitor cells in culture. With some rapidly dividing cell types, such as glia and myoblasts, a large cell population can be derived from the progeny of a single cell. It is therefore possible to have clonal primary cultures, which is the only way to assure that all of the cells in the culture are phenotypically identical. However, clonal primary cells are limited in the number of times that they can divide. This limited life span of cultured normal cells has been associated with in vivo aging. Many of these cell lines display properties of their normal cell counterpart. For example, the v-myc immortalized sympathoadrenal progenitor cell line, MAH, differentiates into neurons upon exposure to fibroblast growth factor. Neuroblastoma C1300 cells can be induced by serum modification to extend neurites.
One problem with the use of clonal cell lines is the limitation in the number of cell lines available that express well-characterized in vivo phenotypes. The derivation from neoplastic tissue suggests an origin which makes the cells abnormal and detracts from their usefulness. However, it needs to be mentioned that all cells acquire abnormal characteristics when placed in a tissue culture environment. Although continuous lines lack the type of growth regulation seen in vivo, the critical point for experimental use is whether the cells express the differentiated characteristic of interest identical to those in vivo. If they do, information on the binding characteristics of the test compound to the target receptor or binding site molecule may be studied as an important first step in the toxicity. However, caution should be exercised prior to using cell lines for toxicity studies even if they express the differentiated characteristic of interest. As the genetic amenability and response to a test compound may be dramatically changed in cell lines as compared to the original primary cells, unforeseen cell line specific effects may occur. Thus, the data may not be applicable for direct extrapolation to the in vivo condition.
One of the major limitations of continuous cell lines is the difficulty in inhibiting cell division experimentally to obtain a stable population of differentiated cells. This is in contrast to the in vivo situation where the end cell in a differentiation sequence usually does not divide. The cellular homogeneity of cell lines may be appropriate for the purpose of studying selectivity of neurotoxicants in toxicological studies. This homogeneity would limit the cellular interactions and the cells would not be exposed to the complex network of messages typical for neuronal circuits. In many cases, differentiation is induced by chemicals and drugs by a mechanism that is not fully understood. It is not known if it is comparable to the differentiation process that occurs in vivo. In general, cell lines show much less sensitivity to toxicants than primary cell cultures. For example, the cholinergic cell line NG-108-15 is much less sensitive to AF64A than primary cells (93).
Genetic stability of any given phenotype is critical to the reproducibility of experimental findings. Clonal cultures can undergo variant selection that can be explained somewhat in terms of point mutations, deletions, or chromosomal rearrangements. Additional data suggest that cells are able to respond in a variety of ways to strong selective agents and that the frequency of variant selection may reflect an interplay between two or more independent mechanisms. Examples of initial phenotypic instability are clonal liver cell lines that frequently lose the ability to synthesize and secrete albumin after a few dozen passages in culture, and skeletal muscle myoblasts that cease to differentiate after a few transfers. Examples of phenotypic change have been reported for PC-12 cells with increased number of passages (94). It is likely that the origin of variants in clonal cells can be explained in terms of classic mutational mechanisms or epigenetic phenomena.
Glioma cell lines. The C6 glioma cell line was derived from a chemically induced glioma in an adult rat (95). These cells possess many of the regulatory control mechanisms and differentiated properties of glial cells. When dibutyryl cyclic AMP is added to primary CNS glia cultures or to glial cell lines, there is an increase in morphological complexity which is reversible upon removal (96). Chemicals which increase intracellular cyclic AMP produce similar morphological changes (97). Dibutyryl cyclic AMP has been used extensively to study the effects of various factors on differentiated glia cells (98). C6 cells have been used in numerous studies both of toxicity and basic cellular mechanisms. This cell line has been established as a model in which several aspects of hormonal action previously observed in vivo or in primary brain cell cultures can be studied (99). C6 glioma cells have been used to study the regulation and modulation of myelin specific genes (100-102). Based upon the in vivo experimental studies and human case reports suggesting that major target sites of lead acetate are myelinating cells of the nervous system, these cells have been used to examine the toxicity of lead acetate. Results suggest that lead has a selective inhibitory effect on an oligodendroglial function expressed in the C6 glial cell line. This was demonstrated by a dose-dependent inhibition of the glucocorticoid induction of soluble cytoplasmic GPDH at the level of transcriptional processing. Since GPDH is a specific biochemical marker for the myelin-forming cells and oligodendrocytes and is believed to be involved in myelination, the selective inhibitory effect of lead on GPDH induction is consistent with the in vivo observations of hypomyelination.
Neuroblastoma cell lines. Cell lines have been established from both mouse and human neuroblastomas which express several neurotransmitter-synthesizing enzymes. This feature makes them good candidates for the study of regulation of gene expression and for the establishment of cDNA libraries. The first neuroblastoma cell lines were derived over 40 years ago. They form homogenous populations, proliferate rapidly in chemically defined media, extend neuritic-like processes and contain several neurotransmitter precursor molecules such as tyrosine hydroxylase. When using neuroblastoma cells it is important, and at times difficult, to distinguish between morphologic differentiation and cytotoxicity (103). In pesticide research, neuroblastoma lines are used because of the presence of the target enzymes AChE and NTE (104). The NIE115 neuroblastoma, cloned from the murine neuroblastoma C1300, has been used to study features associated with organophosphate-induced delayed neuropathy (OPIDN) with an attempt to rank NTE inhibition levels after exposure to various organophosphates (105-107). For these reasons, it has been unofficially proposed as a substitute for the Hen test that the U.S. EPA Pesticide Registry uses to detect organophosphorus pesticides capable of causing neuropathy (OPIDN).
Human neuroblastoma cell lines are similar to mouse or rat neuroblastoma cell lines; they are generated from human tissue and may offer species-specific responses to neurotoxic agents. One such cell line is the IMR32, which differentiates upon stimulation with several drugs and expresses several neuronal characteristics and functions such as neurotransmitter synthesis and storage, receptors, and ion channels. In such cells, the specific neuronal end points examined are associated with neurite outgrowth and expression of neurotransmitter receptors. General cytotoxicity is determined as an indicator of neurotoxic specificity. Pharmacological experiments have demonstrated that IMR32 cells express surface nicotinic and muscarinic cholinergic receptors and that classical pharmacological dynamics are present at these receptor sites. These cells have been used to examine the toxicity of various metals that target the cholinergic system in vivo. SY5Y is a subclone of the human neuroblastoma SK-N-SH and has neurotoxic esterase activity comparable to brain, thus allowing the study of OPIDN-inducing chemicals.
Pheochromocytoma cells. The PC-12 clonal cell line was established from a rat pheochromocytoma (adrenal medullary tumor) and has many properties in common with primary sympathetic neuron and chromaffin cell cultures (94,108). These cells can be induced to differentiate in the presence of NGF to resemble sympathetic neurons, to extend neurites, increase tyrosine hydroxylase (TH) activity, and dedifferentiate with the removal of such growth factors [for review see Fujita et al. (109)]. The PC-12 line has been used extensively to examine the mechanisms underlying NGF response and nerve differentiation. Depending on the conditions, PC-12 cells synthesize AChE, acetylcholine, and choline acetyltransferase as well as release dopamine, norepinephrine, and acetylcholine. They contain sodium, potassium, and calcium channels, and various other membrane receptors including receptors coupled to G-proteins. For these reasons they have been used extensively to examine the basic biology of neurotransmitter biosynthesis and secretion, neuronal differentiation, calcium ionic flux, and signal transduction mechanisms. They provide a useful model for studying processes associated with neuronal differentiation, synthesis, storage, and release of neurotransmitters, function and regulation of ion channels, and interactions of compounds with membrane-bound receptors. In toxicology, PC-12 cells have been used to evaluate chemical induced alterations in calcium channels neurotransmitter release and receptor, and by both biochemical and physiological methods (110-113).
ADDITIONAL CULTURE SYSTEMS. Microcarrier techniques have been developed to study different cell types in vitro (114). These microcarriers have a very large surface-to-volume ratio and can be used to obtain high yields of cells from small culture volumes. This system allows for mixtures of cell types to be produced at any defined ratio and as such offers an attractive model for examining specific questions concerning cell-cell interactions. Microcarrier techniques have been applied to study fiber outgrowth, synaptogenesis, myelin formation in neural cells, and metabolic interactions between cells.
Transgenic mice carrying target constructs of the polyoma large T or
SV40 large T gene, have been used to establish both astroglial and neuronal
cell lines. Recent developments in cell fusion techniques have allowed
neuronal cells of defined origin to be "immortalized" by fusion to a transformed
cell line. For example, the NG108-15 hybridoma was developed by fusion
of the mouse neuroblastoma clone N18TG2 with the rat glioma clone C6BU1
using Sendai virus (115). This hybridoma offers the characteristics of
nerve fiber extension, synapse formation, choline acetyltransferase activity,
and receptors for various neurotransmitters (116). The NSC-34 spinal cord
neuron ![]() neuroblastoma
hybrid cell line has been used to evaluate chemicals that have an in
vivo site of action at the motor neuron (117). These cells have been
produced by fusion of motor neuron-enriched, embryonic mouse spinal cord
cells with mouse N18TG2 neuroblastoma cells (117). The cultures contain
two populations of cells: a) small, undifferentiated cells that
have the capacity to undergo cell division; and b) larger, multinucleate
cells that express many properties of motor neurons. These cells proliferate
in culture, but also express a number of motor neuronlike properties without
the need to add inducing agents like NGF to the culture medium. Such processes
include extension of neurites; generation of action potentials; expression
of neurofilament triplet proteins, neuron-specific enolase and choline
acetyltransferase; synthesis and storage of acetylcholine, induction of
acetylcholine receptor aggregates on co-cultured myotubes; and expression
of a receptor for the neuromuscular junction-specific basal lamina.
neuroblastoma
hybrid cell line has been used to evaluate chemicals that have an in
vivo site of action at the motor neuron (117). These cells have been
produced by fusion of motor neuron-enriched, embryonic mouse spinal cord
cells with mouse N18TG2 neuroblastoma cells (117). The cultures contain
two populations of cells: a) small, undifferentiated cells that
have the capacity to undergo cell division; and b) larger, multinucleate
cells that express many properties of motor neurons. These cells proliferate
in culture, but also express a number of motor neuronlike properties without
the need to add inducing agents like NGF to the culture medium. Such processes
include extension of neurites; generation of action potentials; expression
of neurofilament triplet proteins, neuron-specific enolase and choline
acetyltransferase; synthesis and storage of acetylcholine, induction of
acetylcholine receptor aggregates on co-cultured myotubes; and expression
of a receptor for the neuromuscular junction-specific basal lamina.
The NSC-34 cell responds to agents that affect voltage-gated ion channels, cytoskeletal organization, and some components of axonal transport. Investigation into the use of these cells as a test system for neurotoxicity examined chemicals that exert their neurotoxicity by altering specific neuronal functions: action potential production, axonal transport, neurofilament organization, and neurotransmission (117). The cytoskeletal proteins in NSC-34 cells are sensitive to 2,5-hexanedione in the formation of intermediate filament aggregates. The timing for such studies is critical--as the expression of vimentin decreases with differentiation, the sensitivity of these filaments to aggregation by 2,5-hexanedione decreases (118). Chemical-induced changes linked to axonal transport have been examined only with sodium pyridinethione. In this case, retraction and focal swelling of processes was observed at only the highest dose and may be a manifestation of cytotoxicity rather than an alteration in organelle transport. The cells detected chemical-induced changes in voltage-gated ion channels although there were quantitative differences from primary motor neurons. These cells do not form functional synapses with cultured myotubes and therefore are not a test system for evaluating synaptic transmission (117). Most interesting is the fact that the NSC-34 cell does not require chemical induction to express a differentiated phenotype, providing an advantage for cell biological evaluations. However, the inability to prevent differentiation is also a disadvantage in that the population of cells most likely to differentiate is gradually diminished with successive subculture. Although fusion techniques can create such hybrid cells, many of the neuronal characteristics are lost from such preparations after a certain number of passages. Attempts have been made to transfect primary cells with oncogenes but these cells usually only survive for a short time.
Basal Level of Cell Functioning. Basal level end points reflect generic cell functioning necessary for cell survival. Although basal cell function assays are used as an index of cell lethality, these measurements do not necessarily give an indication of the type of cell death. Measurements of cell toxicity include evaluation of macromolecular synthesis, lipoperoxidation and generation of reactive oxygen species, indicators of mitochondrial and lysosomal activity, loss of ions or cofactors, induction of apoptosis and DNA integrity, energy regulation (oxidative-reduction status), generic cell functions (respiration, ion transport, and protein and DNA turnover), cell-cell communication via gap junction integrity, biosynthetic reactions, transport processes, and specific enzyme changes. Although these processes can be used to assess the status of the cell, basal cell function is often limited to assays such as vital dye uptake, mitochondrial viability assays, total cellular protein, or lactate dehydrogenase release. Vital dye exclusion assays are based on the ability of cells with damaged leaky membranes to allow the entry of stains such as Trypan Blue, or the loss of intracellular stains such as neutral red. Although the vital dye assays are used as an index of cell lethality, what they actually measure is cell membrane permeability. Since cells can survive some degree of membrane permeability, these measurements do not necessarily equate with cell death. In addition, methods such as Trypan blue exclusion require a quantitation of cell numbers containing the dye. The neutral red uptake and 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium (MTT) reduction assays have been widely used as semispecific indices of aspects of cellular function (119). These indices can provide measures of cytotoxic potential that are more sensitive than vital dye exclusion. Any microscopic quantitation can prove to be problematic with cells forming clusters or cells that have a flat morphology such as astrocytes or fibroblasts. With fluorescent dyes such as ethidium homodimer and propidium iodide also excluded from live cells, a fluorescence-activated cell sorter can count the number of fluorescent cells automatically. For this, however, the cells have to be detached from the substratum and/or have to be dissociated into single cells. Another method is to measure total culture fluorescence before and after destruction of the cell membrane. As such assays require washing to remove excess dye, cells that lose adherence or disintegrate would be removed from the final determination.
The cytosolic enzyme LDH has been proposed to monitor toxicant exposures that last for days or weeks. It is assumed that over time the enzyme marker accumulates in the medium and that a stable reading of this accumulation should be available at the end of the exposure. It must be noted that LDH is not a stable enzyme and its half-life can be influenced by other products released by cells. In all cases, any measurement of a substance released by the cells into the medium must be examined relative to the total amount available for release. Therefore, such studies would determine the amount of the marker both in the medium and inside of the cell and express the data as a ratio of released/total marker. One must consider other factors that may come into play, especially in a mixed cellular culture, such as a concomitant induction of endogenous substances. Such substances could degrade proteins in the medium and influence any protein measurement of leaky membranes. Healthy cells uptake such substances as radiolabelled proline, adenosine, and chromium; elevated release can be a reliable and easily monitored index of toxicity. For measurements of 51Cr release, cells are preloaded with 51Cr and the amount released into the medium after toxicant exposure is measured in a gamma counter. The relative amount released as a fraction of the total amount in the preparation is the measure of cytotoxicity. All of these measures reflect the permeability of the membrane and do not directly confirm cell death.
Cytotoxicity and viability end points provide information on the intrinsic toxicity of chemicals. They have limited ability, if any, to predict neural-specific effects. However, such end points must be included to determine the health status of the cells at the time of process evaluation and possibly to differentiate specific effects from general cytotoxicity. In many cases a distinction is made based on the selectivity of the endpoint examined. For example, differentiated cell functions such as axonal transport, synaptogenesis, myelination, enzyme activities, and neurotransmitter function could be examined for neurotoxicity. Characteristics of neurotoxicants could include neurotransmitter-specific accumulation, release, or metabolism by the neural cells. The development of early and cell-specific indicators of the reaction of neuronal cells and astroglial cells to injury will require examination of neuronal and glial-specific protein changes. It is commonly acknowledged that given the complexity of the nervous system, no single endpoint can encompass the range of neurotoxic targets. A suggestions to overcome this complexity has been to include a variety of multiple end points. Again, given the complexity of the nervous system, the ability to select such multiple end points to reflect the dynamics of the nervous system is limited by our current minimal understanding of mechanisms associated with neurotoxicity in vivo.
Target Sites of Neurotoxicity. In the attempt to develop in vitro systems to evaluate the neurotoxic potential of chemicals, one should identify in vivo target sites or processes, understanding the biological details associated with the in vivo process and the types of perturbations that can be induced on any one particular target by various chemicals. A model system cannot be developed in isolation. Without an understanding of the basic biology, the points at which the basic process can be altered, and the types of injury, a model system with any usefulness or meaning cannot be developed. In fact, the development and use of an inappropriate model, whether in vivo or in vitro, can cause damage by providing potentially biologically invalid experimental data. A number of biological processes that are target sites for neurotoxicants in vivo are easily examined with in vitro systems. The following examples will attempt to address some of these proposed processes.
STRUCTURAL. Morphological end points including effects on the cell body, axons and dendrites, can be crucial in the assessment of chemical-induced alterations in target cell populations as different morphologic patterns of chemically induced damage can be observed in vitro (120). Morphologic lesions, however, have received only limited attention as in vitro end points of neurotoxicity. The interpretation of altered morphology in neural cell cultures is hampered by the paucity of descriptions regarding normal cytology, the range of possible cellular lesions, and the types of possible artifacts which may occur.
Neurons exposed to substances in culture often develop structurally abnormal neurites. A common phenomenon is beading, which is the formation of gross organelle-filled dilations spaced at intervals along the neurite. Beading occurs in cells exposed to colchicine (121), acrylamide (122), or ethylene oxide (123). Morphological alterations can be seen in astrocyte cultures in response to chemical agents. For example, trimethyltin alters astrocyte morphology, as demonstrated by a retraction of processes and a central focus and increased size of the cell body (37). Microglia may be induced to differentiate into an ameboid form and initiate phagocytosis (124,37). All neural cells may be induced to change morphological phenotype in the retraction of cellular processes along the substratum.
Neurite Outgrowth. Neurite extension is a fundamental property of neuronal cells in vitro and depends upon a number of critical cellular processes such as axonal transport. Although time-consuming, quantitation of neurite outgrowth is a relatively easy task. In addition, neurite outgrowth can be determined by the measurement of neurite-specific structural elements, for example, microtubule-associated proteins, neurofilament proteins. The ability of biological factors, pharmaceutical agents, or environmental and industrial compounds to interfere with neurite outgrowth is often used to screen activity of new substances. In many cases, one examines a substance's ability to stop ongoing normal outgrowth or the induction of cells to retract neuritic processes. Assays for neurite-promoting factor, neuronotrophic factors, and glial maturation factors are often used to assess effects of exogenous substances on developmental processes. It has been proposed that one may examine a compound's ability to block biologically induced neurite outgrowth as can be seen in nerve growth factor-treated cells (125,126). However, this approach is limited because the inhibition of induced outgrowth may be due to indirect effects resulting from the absence or modification of certain enzymes or structural elements rather than from an effect on the cells' ability to generate neuritic processes. Although this approach may offer information of scientific interest, it may not address the issue of a direct effect of a chemical on neurite outgrowth.
Myelin. One major interaction between neurons and glia that results in a structural component of the nervous system is the process of myelination. There are a number of disease processes and compounds that will selectively alter myelin whether as a hypomyelination, a dysmyelination, or a demyelination [for review see Quarles et al. (127) and Morell (128)]. Myelination is specifically altered by triethyltin and hexachlorophene, by developmental exposure to inorganic lead, and by undernutrition. In vivo research efforts in this area range from morphological examination of myelinated tracts and biochemical evaluation of myelin proteins, to examination of alterations in the regulation of myelin-specific genes.
PERIPHERAL NERVE MYELIN. Because of availability of peripheral nerve tissue, much of the work on processes of myelination both in vivo and in vitro has focused on the myelination of the sciatic nerve by Schwann cells. The difficulties in culturing Schwann cells are similar to other primary neural cultures except for those associated with myelination in culture. Stimulating the cell to myelinate an axon is a very complicated task that has been accomplished by only a few laboratories engaged in mechanism-based research. Even in these cases, the number of myelin lamellae (wrappings) is limited--a significant difference from the in vivo condition. In vivo once Schwann cells are in their final location on the axon, they stop dividing and begin to elaborate myelin around the nerve (129). The presence of myelin along the axon serves to increase the velocity of signal conduction along the nerve and reduces the energy requirement for conducting the impulse. In vivo Schwann cells make galactocerebroside and the myelin-specific protein P0 after they have migrated down the nerves, ceased dividing, and are developmentally committed to myelination (130,131). When they are cultured in the absence of nerve, they initially contain galactocerebroside and P0, but the number of cells containing these molecules declines with a half-life between 2 to 3 days. Given an increase in cell number, this decrease is not due to cell loss but rather a progressive loss of antigens. Much research effort has been directed toward developing a model system for myelination to understand its basic underlying requirements. After 15 to 20 years work, we now know more about the interactions between the myelinating cell of the peripheral nervous system and its environment. However, the culture system is a very demanding system that has to date only been applicable to mechanistic-based research and is beyond the demands of a screening test for neurotoxicity.
CENTRAL NERVOUS SYSTEM MYELIN. Efforts to develop a system to examine CNS myelination by oligodendrocytes have a much shorter and complicated history. For example, unlike the PNS where the relationship between Schwann cell and axon is 1 to 1, the relationship between oligodendrocytes is 1 to 40 separate nerve fibers. In addition, not all Schwann cells and oligodendrocytes produce a myelin sheath. One model recently adapted to examine CNS myelination is the cerebellar slice culture with a modification of substratum attachment rather than a roller bottle method (132). It is still too early to determine whether this model will offer a method to evaluate toxicant-induced alterations in myelin. A model system to evaluate both the process of myelination and structural myelin wrappings would be beneficial in understanding the action of neurotoxicants that selectively target myelin. However, critical in vivo target sites for neurotoxicants are absent, i.e., multiple lamella and nodes of Ranvier (spaces along the axon between each cell's myelin wrapping). For example, triethyltin and hexachlorophene both produce alterations in myelin as evidenced by a splitting of the intraperiod line (the space between myelin lamellae) (133). Without multiple wrappings a similar effect could not be seen in vitro. Chemicals that produce hypomyelination have a better chance of being detected. However, in many of these cases, such as in the developing nervous system after lead exposure, the myelin that is produced is normal in protein and lipid content; there are just fewer lamellae and thus less myelin (134,135). In vivo efforts suggest that the perturbation is on the myelin sheath and not a direct effect upon the myelinating cell. This also would limit the usefulness of a culture system that is restricted to the cell body and to very little if any plasmalemma in the form of myelin.
Although chemicals and disease processes have been classified as primary demyelinating agents, this classification system is based upon a morphological criteria (136). It does not take into account the role of dynamic signaling interactions between the myelin sheath and its underlying axon. Whether the resulting pathology in the myelin sheath is due to a direct effect on myelin or the myelinating cell or in response to a perturbation in the underlying axon is still a major question in whole animal models [for review of neuropathies see Pleasure (137)]. Additional yet critical players to the morphological end product seen in vivo are both the resident and the infiltrating macrophages, which have the role of stripping myelin from the axon and removing it from the nerve area. Any in vitro model system designed to determine effect of a toxicant on myelin would need to include all of the known in vivo active participants.
Existing in vitro systems allow examination of the signaling mechanisms linked to initiation of myelination in the developing organism. However, given the difficulty of the culture, such efforts should be limited to well-defined, focused questions aimed toward understanding the mechanisms associated with perturbations to developing myelinating cells. Efforts have been made to develop an in vitro system to model the vulnerability of the myelin genes in the developing brain. Using mixed glial cultures to preserve the temporal specificity of the myelin gene expression, Royland and co-workers (138) developed a system to examine the effects of nutritional deprivation on myelin gene expression. When the mixed glial cultures were maintained in defined medium, myelin genes responded to various manipulations such as alterations in glucose concentrations. The cultures can be further manipulated to produce cultures of purified oligodendroglia, allowing for more detailed examination of the mechanisms associated with alterations in the expression of specific myelin genes during development. These cell culture systems preserve the in vivo mechanisms of activation and upregulation of myelin genes critical for myelin development in vivo. Thus the primary cultures allow for further examination of toxicant-induced alterations in myelination at the in vitro level and can provide an additional experimental system for studies of normal and abnormal gene expression during development of the brain. Aggregating cell cultures also provide a useful system for biochemical and morphological analysis of myelin and the process of myelination and remyelination (75,139-141). The morphological characteristics in reaggregates are similar to those in vivo, and myelin protein can be isolated in sufficient quantities for biochemical analysis. In addition, the yield of the myelin-associated enzyme CNP and the myelin protein composition is similar between 30-day aggregate cultures and adult rat brain (75).
FUNCTIONAL END POINTS
OF NEUROTOXICITY. Axonal transport.
Axonal transport involves mechanisms similar to those that all eukaryotic
cells use to transport materials from their sites of synthesis to their
sites of utilization and subsequent sites of degradation. Alterations in
this process have broad implications for the biological functioning of
the organism (142). A key question surrounding toxicant-induced alterations
in axonal transport deals with whether the effect occurs by direct or indirect
mechanisms. A toxicant can produce an effect on transport via several mechanisms.
It might interact with a specific step in the mechanism of axonal transport,
directly affect the movement of a specific transport component without
altering transport per se, or alter the spatial arrangement or production
of axonal components. Additionally, an effect might be due to a more general
biochemical disruption such as energy metabolism. The majority of approaches
used to examine toxicant-induced alterations in axonal transport have involved
whole animals. Use of in vitro techniques are now possible with
the establishment of the video-enhanced contrast differential interference
contrast microscopy (VEC-DIC) methodology to examine organelle movement
along the axon. The N1E115 murine neuroblastoma cell line has been used
to study perturbations of differentiation and axonal transport (143-145).
When the cells are exposed to serum-free medium, they extend large, stable
neurites that are well suited to analysis of intraaxonal movement of organelles
by VEC-DIC. Axonal transport is determined by the number of organelles
traversing a line perpendicular to the long axis of the neurite; velocity
is determined by following individual organelles moving along the neurite.
This method has been successful in detecting a significant reduction in
bidirectional organelle flux induced by vinblastine, an antimicrotubule
drug that impairs fast axonal transport in vivo (143). Examination
of acrylamide, a neurotoxicant that affects aspects of axonal transport,
showed a toxicity to differentiating neuroblastoma, but organelle movement
remained unimpaired. Comparison with in vivo results raises the
possibility that rapid transport of radioactive markers could be altered
without impairment of vesicular traffic and lends support to the observation
that a subset of axons or organelles is impaired by acrylamide (146). These
types of defects would not be readily apparent using video microscopy if
the remaining organelles moved normally. Therefore, such an in vitro
system would be limited to detecting perturbations that affected all populations
of axons or neurites. This would be inconsistent with the in vivo
observations of differential vulnerability of sensory or motor nerves to
various neurotoxicants. When these cells were examined for their ability
to detect the toxicity of acrylamide, no alterations in cell morphology
were seen until 2 weeks after exposure to 25 µg/ml acrylamide. In
the exposed state, the growth cone on the leading edge of the outgrowth
process was defective and neurite swellings were noted (147). Similar alterations
in growth cone integrity have been reported in N1E115 cultures, chick clonal
root ganglia cultures, and chick spinal ganglia. Walum et al. (122) showed
that acrylamide exposure of NIE115 cells produced neurite swellings initially
in the distal regions followed by progression to the proximal portion.
This is similar to the distal dying-back axonopathy observed in vivo
following acrylamide exposure. The level of sensitivity of the cells was
determined with comparison of eight cell lines (neuroblastoma 41A3, neuroblastoma
N18, neuroblastoma N1E115, neuroblastoma ![]() glioma hybrid NG108CC15, glioma 13BMG, glioma C6, fibroblast RLF, and fibroblast
RMC). Of all these cells, the N1E115 and NG108CC15 cells, which are considered
the most differentiated of the neuronal cells tested, were found to be
most sensitive based on growth characteristics and morphology.
glioma hybrid NG108CC15, glioma 13BMG, glioma C6, fibroblast RLF, and fibroblast
RMC). Of all these cells, the N1E115 and NG108CC15 cells, which are considered
the most differentiated of the neuronal cells tested, were found to be
most sensitive based on growth characteristics and morphology.
Electrophysiological indices. Receptor and ion channel expression can be studied in vitro using electrophysiological recording, although specific subtypes are best characterized by single-channel recording. Such detailed recording requires cell membranes free from debris, which may be technically difficult to achieve with organotypic or explant cultures. In addition, synaptic function and spontaneous activity in neuronal networks can be readily monitored in any system sufficiently complex to show such activity. The net result of complex functional relationships may be evaluated by relatively simple electrophysiological techniques such as paired pulse inhibition and long-term potentiation.
Blood-Brain Barrier. The blood-brain barrier (BBB) both protects
and nurtures the brain (26,148). The endothelial cells in brain microvessels
are sealed together by tight junctions and this cuff restricts the passage
of most polar and organic molecules from the bloodstream to interstitial
fluid of the brain (149). Molecules pass through the BBB by the aid of
protein and amino acid transport enzymes and carriers such as ![]() -glutamyl
transpeptidase (GGTP), alkaline phosphates, and glucose transported enzyme
(GLUT-1) (150-152). These endothelial cells of the BBB are physically in
contact with astrocytes that influence many of their functions (153-155).
The astrocytes regulate the rigidity of the tight junctions, thus producing
a higher electrical resistance across the cells. They can enhance a variety
of enzyme activities such as GGTP, an amino acid carrier, and regulate
the expression of GLUT-1 protein. Thus, the BBB can be disrupted either
by damaging the endothelial cell or by interfering in the interdependency
of astrocyte and endothelial cells.
-glutamyl
transpeptidase (GGTP), alkaline phosphates, and glucose transported enzyme
(GLUT-1) (150-152). These endothelial cells of the BBB are physically in
contact with astrocytes that influence many of their functions (153-155).
The astrocytes regulate the rigidity of the tight junctions, thus producing
a higher electrical resistance across the cells. They can enhance a variety
of enzyme activities such as GGTP, an amino acid carrier, and regulate
the expression of GLUT-1 protein. Thus, the BBB can be disrupted either
by damaging the endothelial cell or by interfering in the interdependency
of astrocyte and endothelial cells.
Although it has been clearly demonstrated that toxicity is dependent on the sensitivity and developmental stage of target cells, concentration of test compound, and duration of exposure, it has been proposed that any chemical that passes the BBB is neurotoxic (156). Since many chemicals gain access to the brain parenchyma by disrupting the BBB in vivo (157-162) attempts have been made within in vitro neurobiology to develop a model test system. Questions concerning the BBB have focused primarily on the use of isolated microvessels prepared from bovine and rodent tissues (26,153). One recently developed system (163) is a co-culture of a bovine aortic endothelial cell line (or a rat brain endothelial cells) with rat brain astrocytes (or C6 rat glioma cells) in a tridimensional hollow-fiber culturing apparatus. The system is maintained under pulsatile flow conditions to mimic intraluminal blood flow and to allow for chemical delivery. One cell line that has been proposed as a model test system for the BBB is the Madin-Darby canine kidney cell line. This line displays a variety of functional and enzymatic similarities to endothelial cells of the BBB including electrical resistance, tight junctions, and glucose transporter enzymes, offering a number of potential target sites for examination.
Signal Transduction Mechanisms. CALCIUM HOMEOSTASIS. The normal functioning of the neural cell is critically dependent on the intracellular distribution of calcium with cytosolic-free calcium functioning as the critical pool for regulation of intracellular events (164,165). The viability of the cell can be affected when the mechanisms of calcium homeostasis fail (166-168). If this highly synchronous and coordinated process fails, there is an increase in cytosolic-free calcium, which can activate a number of intracellular reactions. Such reactions include the release of neurotransmitter, phosphorylation of proteins, and protease activity (167,169-171). Intracellular calcium also plays a role in the control of neuronal growth and associated growth cone activity. Many neurotoxic agents with diverse mechanisms have been reported to perturb intracellular calcium. Included in these active chemicals are methyl mercury, chlordecone, triethyl lead (172-174), cyanide (175), pyrethroids (176,177), methyl mercury (168,175), PCB congeners (178,179), and aluminum (180-182).
NEUROTRANSMITTERS AND HORMONES. Numerous culture systems have been developed to address specific biological questions with regard to nervous system cellular induction of or responsiveness to neurotransmitters or hormones. With regard to neurotransmitter synthesis, clones fall into four subsets: cholinergic; adrenergic, both cholinergic and adrenergic, and neither neurotransmitter. All four subsets synthesize and secrete large amounts of AChE into the culture medium. Synthesis of both catecholamines and acetylcholine (ACh) tends to increase with morphological differentiation of the cells, although there is no evidence of an exogenous induction of morphological differentiation producing either an increase or decrease in neurotransmitter synthesis. The first clonal cell lines established in culture were derived from C1300 mouse neuroblastoma. Given that this tumor had been passaged for 30 years prior to growing in culture, and that cell lines derived from mouse tumors generally have very unstable karyotypes, the hundreds of clones isolated from the original tumor have widely different characteristics. Neurotransmitter synthesis in C1300 clones is limited largely to ACh and/or catecholamines due to the origin from cells of the neural crest. Cell lines derived from human neuroblastomas also synthesize predominantly ACh and/or catecholamines (183).
Although the numerous mouse-derived neuroblastoma cell lines available could offer a culture system to examine alterations in ACh or catecholamines, they offer very little in other neurotransmitter systems. Rat clonal cell lines offer increased diversity of neurotransmitter systems. These cells make primarily the inhibitory neurotransmitter GABA acid, with a limited number of cells making small amounts of ACh and catecholamines (184). Additional work by Kimes et al. (185) demonstrated that subclones were able to synthesize two or more neurotransmitters, confirming that individual CNS cells, like those from the neural crest, are able to make multiple neurotransmitters. The clonal PC-12 cell line also has the ability to synthesize both ACh and norepinephrine (186). Data on polychlorinated biphenyls suggest an effect on catecholamine metabolism in PC-12 cells similar to CNS dopamine metabolism. This model has been continued to examine the structure-activity relationships among PCBs and catecholamine metabolism. Another group of cultured cells able to synthesize multiple neurotransmitters is the sympathetic ganglia. Dissociated sympathetic cervical ganglia cells from newborn rats are able to synthesize both ACh and catecholamines (187-190).
Cell cultures represent an excellent system in which to study trophic influences of growth factors and other compounds, as the cholinergic neurons are easily assessable for manipulation and observation. For example, cultures of dissociated cells from the septal area of fetal rat brains have been used to study the effects of NGF, thyroid hormones, and gangliosides on septal cholinergic neurons (191). Isolated adrenal medullary chromaffin cells cultured in defined medium are useful for studying the regulation of tyrosine hydroxylase, dopamine ß-hydroxylase and phenylethanolamine-N-methyltransferase, the key enzymes in catecholamine biosynthesis [for reviews see Furshpan et al. (189) and Bunge et al. (192)]. This culture preparation usually require a retrograde perfusion of the intact bovine adrenal gland with collagenase, followed by separation of medulla from cortex, and enzymatic and mechanical dissociation of medullary tissue (193). The ability to induce ACh synthesis is significantly decreased with increased age of the animal from which cells are obtained (188).
The avian ciliary ganglion represents the most studied model of a selected population of cholinergic cells in the peripheral nervous system (194). The development of cholinergic enzymes, the effect of denervation, and the effects of aging on gangliahave been reported (195,196). The avian system has several advantages. Its developmental and aging process is well characterized; its size and location make it easy to dissect; it is comprised of a relatively large and homogeneous cell population and it can be studied both in vivo and in vitro. In the chick, the ciliary ganglion is located 3 to 4 mm from the eye, deep in the orbit, on the branch of the occulomotor nerve that innervates the oblique muscles of the eyeball. In addition to the branch of the occulomotor nerve, ciliary nerves and choroid nerves emerge from the ganglion, providing innervation to the oblique muscles of the eyeball, smooth muscles, and striated muscles of the iris.
Based on immunocytochemical detection of TH and serotonin (5-HT), techniques have been developed for dissection of monoamine nuclei from embryonic brain tissue for dissociated cell cultures, explant cultures, or transplant cultures. Although regions containing TH or 5-HT immunoreactive cells can be delineated as early as embryonic day 12 (E12), it is very difficult to free the neural tube from the surrounding tissue without damage. The earliest stage allowing for dissection is E13. With increasing age, the dissection becomes easier. However, the yield of viable cells decreases rapidly and only one ganglion can be obtained per animal. Thus, the optimal time for tissue collection is E13 or E14 (197). Whitaker-Azmitia and Azmitia (198) reported effects of various serotonergic drugs, drugs of abuse, and other therapeutic agents including antidepressants on a preparation of dissociated primary cells from midbrain regions rich in serotonin neurons. One striking aspect of the data was the variability in the results for any given substance such that the results of uptake of radiolabeled serotonin were presented as qualitative changes rather than a quantitative measurement. Factors contributing to this variability were time in culture, plating density, and presence of steroids or glucose in the media. Based on the effects of amphetamine on brain catecholamine function, NG108-15 neuroblastoma-glioma cells were used to study the involvement of the serotonergic neurotransmitter system in toxicity induced by amphetamine and its various derivatives. [3H]d-Amphetamine uptake and cell viability demonstrated a parallel dose-related decrease after exposure to amphetamine and its various derivatives (199). Cell death as determined by these two measurements could not be blocked by the specific serotonin uptake inhibitor paroxetine, which at higher doses was also toxic to the cells. The ability of the cells to uptake amphetamine and to show a differential pattern of viability response to various derivatives supported the use of this culture system to screen for the neurotoxic effects of drugs of abuse. However, caution is necessary prior to any widespread use of this approach, as the system was unable to detect any differences between the d- and l-isoforms of amphetamine which in vivo have distinctly different effects on the nervous system: the l-isoform has psychoactive properties and the d-isoform lacks such activity.
The sexually dimorphic nucleus of the preoptic area is an example of a morphological gender difference in the rat hypothalamus and has been a valuable model for the study of neural, cellular, and molecular mechanisms by which gonadal steroids influence the nervous system. The nucleus is composed of a heterogeneous population of neurons situated in the medial preoptic area of the brain. The nucleus receives and projects to numerous regions within the limbic system, including connections to most hypothalamic nuclei, and various nuclei within the brain stem (200). This system offers a wealth of characterization on the effects of steroids, the developmental critical period for steroid action and identified markers for neurons of the nucleus. It has been used as a model for the study of hormonal regulation of gene action, which can influence survival, differentiation, and expression of neuropeptides and receptors. This system may offer a model to examine the effects of various estrogenic-like compounds currently of concern for developmental neurotoxicity. Unfortunately, only one tissue punch per region can be obtained for culture from either adult or developing animals.
For the study of fully differentiated neurons, methods have been developed to isolate neurons from the mature mammalian brain. Usually, such methods are used to isolate neurons for electrophysiological measurements and not for culture. It is possible to isolate neurons from small circumscribed regions of the brain which preserve dendritic structure, thus allowing identification of morphological class. Neurons can be isolated with intact synaptic boutons exhibiting spontaneous release of excitatory and inhibitory neurotransmitters (201). The procedure yields approximately 10 to 20% of the original cells and would allow for investigation of specific neurotransmitter-containing neurons.
REACTIVE CHANGES. Reactive changes can be useful end points of toxicity as they can be used to determine threshold concentrations of toxicity or to identify sensitive systems prior to more mechanistic analysis. GFAP is widely used as a marker of toxicant-induced astroglial reaction in vivo and has also shown value in vitro (202). GFAP levels can be increased in the early stages of a response, whereas a decrease is seen with increasing cytotoxicity (203). Such biphasic reactions are common and can lead to problems in interpretation of results. Inhibition of the reduction of MTT has been widely used as a measure of impaired mitochondrial or cytoplasmic reduction capacity, but many agents produce a reactive stimulation of MTT reduction at low doses, followed by a decrease at higher, cytotoxic doses (204). Biphasic responses are also seen in a time-dependent manner. The astrocytic toxin dinitrobenzene decreases the glutathione content of primary astrocytic cultures within 2 hr while at subcytotoxic concentrations a larger reactive increase is seen by 24 hr (205). Therefore, both time and concentration dependency must be established before data interpretation is possible.
Even within common neurotoxic end points, there is considerable variation between chemicals that target a specific process. Current information on sites of action of known neurotoxicants can be helpful in determining the types of target sites to examine. However, such information can be misleading in that it offers the impression that one would be able to determine the potential for neurotoxicity in new and untested chemicals using only current information. Although this may be the case, the techniques to evaluate these target sites are sophisticated and require not only technical training but also a biological understanding of the system in order to conduct meaningful scientific investigation, even at a preliminary level. In neurotoxicology, difficulty arises when attempting to determine which target sites should be used to evaluate neurotoxicity of a chemical with unknown biological properties. Attempts to distinguish between chemicals that produce cytotoxicity versus a selective neurotoxicity in any culture system is hindered by the identification of a suitable global end point for neurotoxicity that can be examined within the culture system of interest. With current advances in cell biology and neurosciences, additional elements of cell processing are being identified which may allow for a limited number of common mechanisms of cellular dysfunction to be identified. Until that time, efforts to understand how neurotoxicants exert an adverse influence on the various cells of the nervous system will continue, based upon current knowledge of neurobiological functioning, in vivo analyses, previously identified sites of action for numerous chemical substances, and a great deal of hard work.
The other definition refers to the validation of the end point. In this context, one determines whether the test can measure or predict what it is designed to measure, and whether the end point is mechanistically related to the effect of interest, including criteria for interpretation of results. Some models may be biologically useful but not predictive. In this process one would need to use analogs that are active and inactive with respect to process, and examine effects in context of general toxicity.
Because multiple samples can be generated from any one specific source, whether a primary culture, cell line, or brain slice, it is critical that each distinct preparation be considered as the statistical unit (i.e., n for statistical analysis). For example, even though a number of brain slices can be obtained from any given animal, the source remains one animal and the animal remains the statististical unit. The same is true for a primary culture requiring a pooled number of animals. The individually plated wells all represent one preparation, an n of 1. Within such preparations, it is usually considered good experimental protocol to assay each component of the evaluation in triplicate. Such procedures are standard in most biochemical assays.
The description of a dose-response curve obtained from in vitro experiments is generally based on a linear regression analysis, plotting the logarithm of the dose on the horizontal axis and the response on the vertical axis. Although this method allows for calculation of an effective dose, it does not allow for examination of the slope of the dose-response curve. Each of these values is important and can be manipulated by the choice of data points. Such linear regression analysis can not describe complex dose-response curves such as those showing a biphasic response. For a review of mathematical approaches to determine dose-response relationships in vitro, see Bruinink (32).
Veronesi (211) identified the need for a variety of cell culture models and multidisciplinary end points to parallel neurotoxic targets in vivo. This can be difficult because few specific targets of neurotoxic action are known. To differentiate neurotoxicants from other chemicals, it was recommended that nonneural cells be included in any battery to index basal-level functioning. To accomplish this task a tiered approach was proposed. The first tier would include test systems to differentiate neurotoxicants from cytotoxicants. However, even this stage would require using combinations of nonspecific and specific neural end points and batteries of neural and nonneural cell lines. The second tier would differentiate classes of neurotoxicants (metals, cholinesterase inhibitors, solvents, etc.) using specific end points that are specifically targeted in vivo. This could be difficult, given the variability of target site relative to age and length of exposure. For example, at high doses, carbon disulfide is acutely toxic to the CNS whereas lower doses and prolonged exposures produce a peripheral neuropathy. The third tier would address distinct questions confined to specific classes of neurotoxicants e.g., structure-activity relationships.
Preliminary evaluations of a similar screening protocol have been published (212). Using selected chemicals and a range of systems and simple end points, a reasonable correlation between in vitro and in vivo toxicity was seen. However, 2 of 21 chemicals gave a false negative, and 4 positives gave an incorrect target cell type (astrocytes, not neurons). This result illustrates both the potential uses and actual limitations of in vitro systems as open-ended screening systems.
Many of the in vitro experiments that assess the toxicity of various chemicals and drugs demonstrate the difficulty of correlating in vivo and in vitro effects of neurotoxicants and the limitations of an in vitro model as an alternative approach for investigating complex effects at the level of the nervous system. However, they present an opportunity to use in vitro methodologies for studying the underlying mechanisms of action of toxicants. The information obtained from such studies can be used in the refinement of future studies in vivo. The use of cells in vitro has resulted in new insights into mechanisms potentially important in our eventual understanding of the mechanisms of action of neurotoxicants during both development and aging of the nervous system and degenerative brain disorders. Validation should include analysis of the biomechanisms at work in the model system and comparison with the much more complex inner life of the target. Usually the scope of such a comparison is limited. Therefore, we are forced to limit the validation process to a comparison of the outcome of the model experiments with the results of a comparable challenge of the particular biological system. Maintaining the requirements for determining if a chemical is neurotoxic with in vivo data from, the question remains whether in vitro systems can be developed that will be able to make the same determination.
It is important that procedures used in in vitro neurotoxicology are valid. In this context, test methods should both measure what they are designed to measure, i.e., construct validity, and have the ability to measure a characteristic relative to some generally recognized standard, i.e., criterion validity. The results of in vitro procedures should also be interpretable within the context of plausible biological responsiveness to toxicological exposure and possess the ability to predict neurotoxic risk in humans. The process of demonstrating predictive validity can be facilitated by selecting in vitro end points associated with known biological mechanisms of neurotoxic effect. Therefore, attempts to validate in vitro tests for neurotoxicity testing should be limited to test systems designed for and based on assessment of specific mechanisms of neurotoxicity. The sole use of open-ended general toxicity in vitro screens of neurotoxicity at this time does not seem warranted.
In vitro techniques have their greatest potential in experiments involving mechanistically based hypothesis testing. In vitro approaches could be used in several target sites where there is a significant understanding of basic biological processes including chemically induced effects on receptor-mediated changes, neurite outgrowth, electrophysiological changes, certain neurochemical end points (e.g., neuropathic esterase/acetylcholinesterase, ion channel function), and alterations in specific neurotransmitters, enzymes and/or hormones. In vitro procedures may be particularly useful in assessing questions concerning structure-activity relationships. Other potential target sites for in vitro examination include axonal transport, blood-brain barrier, growth and development, astrocytic-neuron interactions, bioactivation, free radical/antioxidant systems, xenobiotic metabolic capacity, and myelination. In each case, the choice of the culture preparations and complexity of the in vitro system should be appropriate for the hypothesis following additional methods development and validation tested.
References
1. U.S. Environmental Protection Agency. Final Report. Principles of Neurotoxicity Risk Assessment [Notice]. Fed Reg 59:42360-42404 (1994).
2. National Academy of Sciences. Risk Assessment in the Federal Government: Managing the Process. Washington:National Academy Press, 1983.
3. National Research Council. Science and Judgment in Risk Assessment. National Research Council, Washington:National Academy Press, 1994.
4. Arenander AT, de Vellis J. Frontiers of glial physiology. In: The Clinical Neurosciences (Rosenberg R, ed). New York:Churchill Livingstone, 1983;53-91.
5. Fedoroff S, Vernadakis A. Astrocytes. Vol 2. Orlando, FL:Academic Press, 1987.
6. Saneto RP, de Vellis J. Neuronal and glial cells: cell culture of the central nervous system. In: Neurochemistry--A Practical Approach (Turner AJ, Bachelard HS, eds). Washington:IRL Press, 1987:27-63.
7. Shahar A, de Vellis J, Vernadakis A, Haber B. A Dissection and Tissue Culture Manual of the Nervous System. New York:Alan R. Liss, 1989.
8. Jacobson M. Developmental Neurobiology. New York: Plenum Press, 1991.
9. Fedoroff S. In vitro systems in medical research. In: The Future of Animals, Cells, Models, and Systems in Research, Development, Education, and Testing. Washington:National Academy of Sciences, 1977.
10. Juurlink BH, Hertz L. Plasticity of astrocytes in primary cultures: an experimental tool and a reason for methological caution. Dev Neurosci 7:263-277 (1985).
11. U.S. Environmental Protection Agency. Proposed Guidelines for Neurotoxicity Risk Assessment [Notice]. Fed Reg 60: 52032-52056 (1995).
12. Nardone RM. Toxicity testing in vitro. In: Growth, Nutrition, and Metabolism of Cells in Culture, Vol III (Rothblatt GH, Cristofalo VJ, eds). New York:Academic Press, 1977;471-495.
13. Rees KR. Cells in culture in toxicity testing: a review. J R Soc Med 73:261-264 (1980).
14. Schrier BK. Nervous system cultures as toxicologic test systems. In: Target Organ Toxicology Series: Nervous System Toxicology (Mitchell CL, ed). New York:Raven Press, 1982;337-348.
15. Balls M, Clothier R. Differentiated cell and organ culture in toxicity testing. Acta Pharm Toxicol 52(Suppl II):115-137 (1983).
16. Grisham JW, Smith GJ. Predictive and mechanistic evaluation of toxic responses in mammalian cell culture systems. Pharmacol Rev 36:151s-171s (1984).
17. Purchase IFH, Conning DM. International Conference on Practical in Vitro Toxicology. Food Chem Toxicol 24:447-818 (1986).
18. Moriuchi S, Shimizu K, Miyao Y, Yamada M, Ohkawa M, Hayakawa T. In vitro assessment for neurotoxicity of antitumor agents before local administration into central nervous system. Anticancer Res 16:135-140 (1996).
19. Vickers PJ, O'Neill GP, Mancini JA, Charleson S, Abramovitz M. Cross-species comparison of 5-lipoxygenase-activating protein. Mol Pharmacol 42:1014-1019 (1992).
20. Lowndes HE, Beiswanger CM, Philbert MA, Reuhl KR. Substrates for neural metabolism of xenobiotics in adult and developing brain. Neurotoxicology 15:61-73 (1994).
21. Kohn J, Durham HD. S9 liver fraction is cytotoxic to neurons in dissociated cultures. Neurotoxicology 14:381-386 (1993).
22. Ransom BR, Kunis DM, Irwin I, Langston JW. Astrocytes convert the Parkinsonism inducing neurotoxin, MPTP, to its active metabolite MPPT. Neurosci Lett 75:323-328 (1987).
23. Hamberger A, Nystrom B, Sellstrom A, Woiler CT. Amino acid transport in isolated neurons and glia. Adv Exp Med Biol 69:221-236 (1976).
24. Holtzman D, DeVries C, Nguyen H, Olson J, Bensch K. Maturation of resistance to lead encephalopathy: cellular and subcellular mechanisms. Neurotoxicology 5:97-124 (1984).
25. Bruinink A. Serum-free monolayer cultures of embryonic chick brain and retina: immunoassays of developmental markers, mathematical data analysis and establishment of optimal culture conditions. In: The Brain in Bits and Pieces (Zbinden G, ed). Verlag Zolikon, 1992;23-50.
26. Betz AL, Goldstein GW, Katzman R. Blood-brain-cerebrospinal fluid barriers. In: Basic Neurochemistry, 5th ed (Siegel GJ, Agranoff BW, Albers RW, Molinoff PB, eds). New York:Raven Press, 1994;681-701.
27. Ronnett GV, Hester LD, Nye JS, Connors K, Snyder SH. Human cortical neuronal cell line: establishment from a patient with unilateral megalencephaly. Science 248:603-605 (1990).
28. Stewart RM, Rosenberg RN. Physiology of glia: glial-neuronal interactions. Int Rev Neurobiol 21:275-309 (1979).
29. Hatten ME. Neuronal regulation of astroglial proliferation and differentiation. In: The Assembly of the Nervous System. New York:Alan R. Liss, 1989;151-166.
30. Hatten ME. Neuronal regulation of astroglia morphology and proliferation in vitro. J Cell Biol 100:384-396 (1985).
31. Hatten ME. Neuronal regulation of astroglial proliferation is membrane-mediated. J Cell Biol 104:1353-1360 (1987).
32. Bruinink A, Reiser P, Muller M, Gahwiler BH, Zbinden G. Neurotoxic effects of bismuth in vitro. Toxicol In Vitro 6:285-293 (1992).
33. Hatten ME, Shelanski ML. Mouse cerebellar granule neurons arrest the proliferation of human and rodent astrocytoma cells in vitro. J Neurosci 8:1447-1453 (1988).
34. Laterra J, Bressler JP, Indurti RR, Belloni-Olivi L, Goldstein GW. Inhibition of astroglia-induced endothelial differentiation by inorganic lead: a role for protein kinase C. Proc Natl Acad Sci USA 89:10748-10752 (1992).
35. Richter-Landsberg, Besser A. Effects of organotins on rat brain astrocytes in culture. J Neurochem 63:2202-2209 (1994).
36. Thompson, TA, Lewis JM, Dejneka S, Severs WB, Polavarapu R, Billingsley ML. Induction of apoptosis by organotin compounds in vitro: neuronal protection in antisense oligonucleotides directed against stannin. J Pharmacol Exp Ther 276:1201-1215 (1996).
37. Maier WE, Bartenbach MJ, Brown HA, Tilson HA, Harry GJ. Induction of tumor necrosis factor alpha in cultured glial cells by trimethyltin. Neurochem Int 30:385-392 (1997).
38. McCarthy KD, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol 85:890-902 (1980).
39. Koelle GB. The histochemical differentiation of types of cholinesterases and their localizations in tissues of the cat. J Pharmacol Exp Ther 100:158-179 (1950).
40. Henn F, Hamberger A. Glial cell function: uptake of transmitter substances. Proc Natl Acad Sci USA 68:2686-2690 (1971).
41. Schousboe A, Hertz L. Regulation of glutamatergic and GABAergic neuronal activity by astroglial cells. In: Dale's Principle and Communication Between Neurones (Osborne NO, ed). Oxford:Pergamon Press, 1984;113-141.
42. Desmedt JE, LaGrutta G. The effect of selective inhibition of pseudocholinesterase on the spontaneous and evoked activity of the cat's cerebral cortex. J Physiol 136:20-40 (1957).
43. Bowery NG, Brown DA, Collins CGS, Galvan M, Marsh S, Yamani G. Indirect effects of amino-acids on sympathetic ganglion cells mediated through release of *-aminobutyric acid from glial cells. Br J Pharmacol 57:73-91 (1976).
44. Schousboe A, Larsson OM, Frandsen A, Belhage B, Pasantes-Morales H, Krogsgaard-Larsen P. Neuromodulatory actions of glutamate, GABA, and taurine: regulatory role of astrocytes. In: Plasticity and Regeneration of the Nervous System (Timeras PS, Privat A, Giacobini E, Lauder J, Vernadakis A, eds). New York:Plenum Press, 1991;165-180.
45. Teichberg VI. Glial glutamate receptors: likely actors in brain signaling. FASEB J 5:3086-3091 (1991).
46. Martinez-Hernandez AM, Bell KP, Norenberg MD. Glutamine synthetase: glial localization in brain. Science 195:1356-1358 (1977).
47. Cammer W. Glutamine synthetase in the central nervous system is not confined to astrocytes. J Neuroimmunol 26:173-178 (1990).
48. Ericinska M, Silver IA. Metabolism and role of glutamate in mammalian brain. Prog Neurobiol 35:245-296 (1990).
49. Flott B, Seifert W. Characterization of glutamate uptake systems in astrocyte primary cultures from rat brain. Glia 4:293-304 (1991)
50. Gallo V, Giovanni C, Suergiu R, Levi G. Expression of excitatory amino acid receptors by cerebellar cells of the type 2 astrocyte cell lineage. J Neurochem 52:1-9 (1989).
51. Kaplan GP, Hartman BK, Creveling CR. Immunohistochemical demonstration of catechol-O-methyltransferase in mammalian brain. Brain Res 167:241-250 (1979).
52. Levitt P, Pintar JE, Breakefield XO. Immunocytochemical demonstration of monoamine oxidase B in brain astrocytes and serotonergic neurons. Proc Natl Acad Sci USA 79:6385-6389 (1962).
53. Westlund KN, Denny RM, Kochersperger LM, Rose RM, Abell CW. Distinct monoamine oxidase A and B populations in primate brain. Science 230:181-183 (1985).
54. Tiffany-Castiglioni E. Cell culture models for lead toxicity in neuronal and glial cells. Neurotoxicology 14:367-390 (1993).
55. Tiffany-Castiglioni E, Zmudzki J, Wu JN, Rowles TK. Effects of lead treatment on intracellular iron and copper concentrations in cultured astroglia. Metab Brain Res 2:61-79 (1987).
56. Wu JN, Tiffany-Castiglioni E. Reduction by lead of hydrocortisone-induced glycerolphosphate dehydrogenase activity in cultured rat oligodendroglia. In vitro. Cell Dev Biol 23:765-774 (1987).
57. Gebhart AM, Goldstein GW. Use of in vitro system to study the effects of lead on astrocyte-endothelial interactions: a model for studying toxic injury to the blood-brain barrier. Toxicol Appl Pharmacol 94:191-206 (1988).
58. Choi DW, Rothman SM. The role of glutamate neurotoxicity in hypoxic ischemia neuronal death. Annu Rev Neurosci 13:171-182 (1990).
59. Ray DE. Pesticides derived from plants and other organisms. In: Handbook of Pesticide Toxicology (Hayes WJ, ed). New York:Academic Press, 1991;585-636.
60. Bottenstein JE, Sato G. Cell Culture and the Neurosciences. New York:Plenum Press, 1985.
61. Bruinink A, Reiser P. Ontogeny of MAP2 and GFAP antigens in primary cultures of embryonic chick brain. Effect of substratum, oxygen tension, serum and Ara-C. Int J Dev Neurosci 9(3):269-279 (1991).
62. Smith RA. Primary cultures of adult mammalian sensory neurons and other in vitro systems of use in neurotoxicological studies. Arch Toxicol Suppl 14:8-14 (1991).
63. Smith RA, Jiang Z-G. Neuronal moduation and plasticity in vitro. Int Rev Cytol 153:233-296 (1994).
64. Dawis S, Hofmann H, Niemeyer G. The electroretinogram, standing potential, and light peak of the perfused cat eye during acid-base changes. Vision Res 25:1163-1177 (1985).
65. Schneider T, Zrenner E. The effect of fluphenazine on rod-mediated retinal responses. Doc Ophthalmol 65:287-296 (1987).
66. Linnemann D, Lyles JM, Bock E. A developmental study of the biosynthesis of the neural cell adhesion molecule. Dev Neurosci 7:230-238 (1985).
67. Teyler TJ. The brain slice preparation: hippocampus. Brain Res Bull 5:391-403 (1980).
68. Rimvall K, Keller F, Waser PG. Development of cholinergic projections in organotypic cultures of rat septum, hippocampus and cerebellum. Dev Brain Res 19:267-278 (1985).
69. Fountain SB, Teyler TJ. Characterizing neurotoxicity using the in vitro hippocampal brain slice preparation: heavy metals. In: Model Systems in Neurotoxicological approaches to Animal Testing. New York:Alan R. Liss, 1987;19-31.
70. Seeds NW. Differentiation of aggregating brain cell cultures. In: Tissue Culture of the Nervous System (Sato G, ed). New York:Plenum Press, 1973;35-53.
71. Honegger P, Richelson E. Biochemical differentiation of mechanically dissociated mammalian brain in aggregating cell culture. Brain Res 109:335-354 (1976).
72. Honegger P. Biochemical differentiation in serum-free aggregating brain cell cultures. In: Cell Culture in the Neurosciences (Bottenstein JE, Sato G, eds). New York:Plenum Press, 1985;223-243.
73. DeLong GR. Histogenesis of fetal mouse isocortex and hippocampus in reaggregating cell cultures. Dev Biol 22:563-583 (1970).
74. Lu EJ, Brown WJ, Cole R, deVellis J. Ultrastructural differentiation and synaptogenesis in aggregating rotation cultures of rat cerebral cells. J Neurosci Res 5:447-463 (1980).
75. Trapp BD, Honneger P, Richelson E, Webster H deF. Morphological differentiation of mechanically dissociated fetal rat brain in aggregating cell cultures. Brain Res 160:117-180 (1979).
76. Seeds NW, Vatter AE. Synaptogenesis in reaggregating brain cell cultures. Proc Natl Acad Sci USA 68:3219-3222 (1971).
77. Trapp BD, Webster H deF, Johnson D, Quarles RH, Cohen SR, Murray MR. Myelin formation in rotation-mediated aggregating cell cultures: immunocytochemical, electronmicroscopic and biochemical observations. J Neurosci 2:986-993 (1982).
78. Honegger P, Werffeli P. Use of aggregating cell cultures for toxicological studies. Experientia 44:817-823 (1988).
79. Schilter B, Honegger P. Validation of three in vitro toxicity/ teratogenicity test systems using identical coded compounds. I. Aggregating brain cell cultures. Teratology 44:31A (1991).
80. Davenport CJ, Williams DA, Morgan KT. Neurotoxicology using cell culture, CIIT Activities 9:1 (1989).
81. Brockes JP, Fields KL, Raff MC, Studies on cultured rat Schwann cells. I. Establishment of purified populations from cultures of peripheral nerve. Brain Res 165:105-118 (1979).
82. Mason PW, Attema BL, DeVries GH. Isolation and characterization of Schwann cells from frozen sciatic nerves. Transact Am Soc Neurochem 22 (1991).
83. Tennekoon GI, Yoshino J, Peden KWC, Bigbee J, Rutkowski JL, Kishimoto Y, DeVries G, McKhann GM. Transfection of neonatal rat Schwann cells with SV-40 large T antigen gene under control of the metallothionein promoter. J Cell Biol 105:2315-2325 (1987).
84. Yoshino JE, Neuberger TJ, Cornbrooks CJ, Tennekoon GI, Eng LF, DeVries GH. Proliferation and differentiation of a transfected Schwann cell line is altered by an artificial basement membrane. Glia 3:315-321 (1990).
85. McCarthy K, Partlow L. Neuronal stimulation of 3H-thymidine incorporation by primary cultures of highly purified non-neuronal cells. Brain Res 114:415-426 (1976).
86. Salzer JL, Bunge RP. Studies of Schwann cell proliferation. I: An analysis in tissue culture of proliferation during development, Wallerian degeneration and direct injury. J Cell Biol 84:739-752 (1980).
87. Salzer JL, Williams AK, Glaser L, Bunge RP. Studies of Schwann cell proliferation. II: Characterization of the stimulation and specificity of the response to neurite membrane fraction. J Cell Biol 84:753-766 (1980).
88. Salzer JL, Bunge RP, Glaser L. Studies of Schwann cell proliferation. III: Evidence for the surface localization of the neurite mitogen. J Cell Biol 84:767-778 (1980).
89. DeVries GH, Salzer JL, Bunge RP. Axolemma-enriched fractions isolated from PNS and CNS are mitogenic for cultured Schwann cells. Dev Brain Res 3:295-299 (1982).
90. Yoshino JE, Dinneen MP, Lewis BL, Meador-Woodruff JH, DeVries GH. Differential proliferative responses of cultured Schwann cell to axolemma and myelin-enriched fractions. J Cell Biol 99:2309-2323 (1984).
91. Pleasure D, Kreider B, Shuman S, Sobue G. Tissue culture studies of Schwann cell proliferation and differentiation. Dev Neurosci 7:364-373 (1985).
92. Diamond L, Baird WM. Chemical carcinogenesis in vitro. In: Growth, Nutrition and Metabolism of Cells in Culture, Vol 3 (Rothblat GH, Cristofalo VJ, eds). New York:Academic Press, 1977;421.
93. Sandberg K, Schnaar RL, McKinney M, Fisher A, Coyle JT. AF64A: an active site directed irreversible inhibitor of acetylcholinetransferase. J Neurochem 44:439-445 (1985).
94. Guroff G. PC12 cells as model of neuronal differentiation. In: Cell Culture in the Neurosciences (Bottenstein JE, Sato G, eds). New York:Plenum Publishing, 1985;245-272.
95. Benda P, Lightbody J, Sato G, Levine l, Sweet W. Differentiated rat glial cell strain in tissue culture. Science 161:370-371 (1968).
96. Kanje M, Walum E, Edstrom A. Effects of dibutyryl cyclic AMP and cytochalasin B on cultured human glioma cells. Z Mikrosk Anat Frosch 93:487-496 (1979).
97. Edstrom A, Kanje M, Lofgren P, Walum E. Drug-induced alterations in morphology and level of cAMP in cultured human glioma cells. Exp Cell Res 95:359-364 (1975).
98. Fontana A, Grieder A, Arrenbrecht S, Grob P. In vitro stimulation of glia cells by a lymphocyte-produced factor. J Neurol Sci 46:55-62 (1980).
99. Kumar S, Sachar K, Huber J, Weingarten DP, de Vellis J. Glucocorticoids regulate the transcription of glycerol phosphate dehydrogenase in cultured glial cells. J Biol Chem 260:14743-14747.
100. Ye P, Kanoh M, Zhu W, Laszkiewicz I, Royland JE, Wiggins RC, Konat G. Cyclic AMP-induced upregulation of proteolipid protein and myelin associated glycoprotein gene expression in C6 cells. J Neurosci Res 31:578-583 (1992).
101. Zhu W, Kanoh M, Ye P, Laszkiewicz I, Royland JE, Wiggins RC, Konat G. Retinoic acid-regulated expression of proteolipid protein and myelin-associated glycoprotein genes in C6 glioma cells. J Neurosci Res 31:745-750 (1992).
102. Grubinska B, Laszkiewicz I, Royland J, Wiggins RC, Konat GW. Differentiation-specific demethylation of myelin associated glycoprotein gene in cultured oligodendrocytes. J Neurosci Res 39:233-242 (1994).
103. Erkell LJ, Walum E. Differentiation of cultured neuroblastoma cells by urea derivatives. FEBS Lett 104:401-404 (1970).
104. Carrington CD, Carrington MN, Abou-Donia MB. Neurotoxic esterase in cultured cells: an in vitro alternative for the study of organophosphorus compound-induced delayed neurotoxicity. In: Alternative Methods in Toxicology. Vol 3: In Vitro Toxicology (Goldberg AM, ed). New York:Mary Ann Liebert, 1985;457-465.
105. Fedalei A, Nardone RL. An in vitro alternative for testing the effect of organophosphates on neurotoxic esterase activity. In: Alternative Methods in Toxicology. Vol I: Product Safety Evaluation (Goldberg AM, ed). 1983;252-267.
106. Atterwill CK, Johnston H, Thomas SM. Models for the in vitro assessment of neurotoxicity in the nervous system in relation to xenobiotic and neurotrophic factor-mediated events. Neurotoxicology 13:39-53 (1992).
107. Veronesi B, Ehrich M. Differential cytotoxic sensitivity in mouse and human cell lines exposed to organophosphate insecticides. Toxicol Appl Pharmacol 120:240-246 (1993).
108. Greene LA, Tischler AS. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci USA 73:2424-2428 (1976).
109. Fujita K, Lazaroxici P, Guroff G. Regulation of the differentiation of PC12 pheochromocytoma cells. Environ Health Perspect 80:127-142 (1989).
110. Sandberg K, Berry CJ, Rogers TB. Studies on the intoxication pathway of teatanus toxin in the rat pheochromocytoma (PC12) cell line. Binding, internalization, and inhibition of acetylcholine release. J Biol Chem 264:5679-5686 (1989).
111. Seegal RF, Brosch K, Bush B, Ritz M, Shain W. Effects of Arochlor 1254 on dopamine and norepinephrine concentrations in pheochromocytoma (PC-12) cells. Neurotoxicology 10:757-764 (1989).
112. Shafer TJ, Contreras ML, Atchison WD. Characterization of interactions of methyl-mercury with Ca2+ channels in synaptosomes and pheochromocytoma cells: radiotracer flux and binding studies. Mol Pharmacol 38:102-113 (1990).
113. Shafer TJ, Atchison WD. Methylmercury blocks N- and L-type Ca2+ channels in nerve growth factor-differentiated pheochromocytoma cells. J Pharmacol Exp Ther 258:149-157 (1991).
114. Westergaard N, Sonnewald U, Petersen SB, Schousbe A. Characterization of microcarrier cultures of neurons and astrocytes from cerebral cortex and cerebellum. Neurochem Res 16:919-923 (1991).
115. Hamprech B. Structural, electrophysiological, biochemical, and pharmacological properties of neuroblastoma-glioma cell hybrids in culture. Int Rev Cytol 49:99-170 (1977).
116. Dahms NM, Schnaar RL. Ganglioside composition is regulated during differentiation in the neuroblastoma X glioma hybrid cell line NB 1-8-115. J Neurosci 3:806-817 (1983).
117. Durham HD, Dahrouge S, Cashman S. Evaluation of the spinal cord neuron X neuroblastoma hybrid cell line NSC-34 as a model for neurotoxicity testing. Neurotoxicology 14:387-396 (1993).
118. Durham HD. 2,5-Hexanedione aggregates vimentin, but not keratin-intermediate filaments of PtK1 cells. Cell Biol Int Rep 11:307-404 (1987).
119. Atterwill CK, Walum E. Neurotoxicity in vitro: model systems and practical applications. Toxicol In Vitro 3:159-161 (1989).
120. Bolon B, Dorman DC, Bonnefoi MS, Randall H, Morgan KT. Histopathologic approaches to chemical toxicity using primary dissociated neural cells grown in chamber slides. Toxicol Pathol 21:465-479 (1993).
121. Takenaka T. Particle movement in axonal transport. In: Axoplasmic Transport (Iqbal Z, ed). Boca Raton, FL:CRC Press, 1886;109-118.
122. Walum E, Ekblad-Sekund G, Nyberg E, Gustafsson L. Cultured neuroblastoma cells as neurotoxicological models: acrylamide induced neurite disintegration. In: Model Systems in Neurotoxicology: Alternative Approaches to Animal Testing (Shahar A, Goldberg AM, eds). New York:Alan R. Liss, 1987;121-136.
123. Garnass KR, Windebank AJ, Blexrud MD, Kurtz SB. Ultrastructural changes produced in dorsal root ganglia in vitro by exposure to ethylene oxide from hemodialyzers. J Neuropathol Exp Neurol 50:256-262 (1991).
124. Monnet-Tschudi F, Zurich MG, Pithon E, van-Melle G, Honegger P. Microglial responsiveness as a sensitive marker for trimethyltin (TMT) neurotoxicity. Brain Res 690:8-14 (1995).
125. Campenot RB. Local control of neurite development by nerve growth factor. Proc Natl Acad Sci USA 74:4516-4519 (1977).
126. Luckenbill-Edds L, Van Horn C, Greene LA. Fine structure of initial outgrowth of processes induced in a pheochromocytoma cell line (PC12) by nerve growth factor. J Neurocytol 8:493-511 (1979).
127. Quarles RH, Morell P, McFarlin DE. Diseases involving myelin. In: Basic Neurochemistry, 5th ed (Siegel GJ, Agranoff BW, Albers RW, Molinoff PB, eds). New York:Raven Press, 1994;771-792.
128. Morell P. Myelin. New York:Plenum Press, 1984.
129. Webster HF. Development of peripheral myelinated and unmyelinated nerve fibers. In: Peripheral Neuropathology Vol 1 (Dyck PJ, Thomas PK, Lambert EH, eds). Philadelphia:WB Saunders, 1975;37-61.
130. Wood JG, Engel EL. Peripheral nerve glycoproteins and myelin fine structure during development of the rat sciatic nerve. J Neurocytol 5:605-615 (1976).
131. Willison HJ, Ilyas AI, O'Shannessy DJ, Pulley MJ, Trapp BD, Quarles RH. Myelin associated glycoprotein and related glycoconjugates in developing cat peripheral nerve: a correlative biochemical and morphometric study. J Neurochem 49:1853-1862 (1987).
132. Notterpek LM, Bullock PN, Maled-Hedayat S, Fisher R, Rome LH. Myelination in cerebellar slice cultures: development of a system amenable to biochemical analysis. J Neurosci Res 36:621-634 (1993).
133. Towfighi J. Hexachlorophene. In: Experimental and Clinical Neurotoxicology (Spencer PS, Schaumburg HH, eds). Baltimore:Williams & Wilkins, 1980;440-455
134. Krigman MR, Bouldin TW, Mushak P. Lead. In: Experimental and Clinical Neurotoxicology (Spencer PS, Schaumburg HH, eds). Baltimore:Williams & Wilkins, 1980;490-507.
135. Cammer W. Toxic demyelination: biochemical studies and hypothetical mechanisms. In: Experimental and Clinical Neurotoxicology (Spencer PS, Schaumburg HH, eds). Baltimore:Williams & Wilkins, 1980;92-99.
136. Spencer PS, Schaumburg HH. Classification of neurotoxic disease: a morphological approach. In: Experimental and Clinical Neurotoxicology (Spencer PS, Schaumburg HH, eds). Baltimore:Williams & Wilkins, 1980;92-99.
137. Pleasure DE. Biochemistry of neuropathy. In: Basic Neurochemistry, 5th ed (Siegel GJ, Agranoff BW, Albers RW, Molinoff PB, eds). New York:Raven Press, 1994;761-770.
138. Royland JE, Konat GW, Wiggins RC. Myelin gene activation; a glucose sensitive critical period in development. J Neurosci Res 36:399-404 (1993).
139. Matthieu JM, Honegger P, Farrod P, Gautier E, Dolivo M. Biochemical characterization of a myelin fraction isolated from rat brain aggregating cell cultures. J Neurochem 32:869-881 (1979).
140. Matthieu JM, Comte V, Tosic M, Honegger P. Myelin gene expression during demyelination and remyelination in aggregating brain cell cultures. J Neuroimmunol 40:231-234 (1992).
141. Zurich MG, Matthieu JM, Honegger P. Improvement of culture conditions for aggregating brain cells to study remyelination. Schweiz Arch Neurol Psychiatr 144:228-232 (1993).
142. Goodrum JF, and Morell P. Axonal transport. In: Neurotoxicology (Abou-Donia MB, ed). Boca Raton, FL:CRC Press, 1992;79-120.
143. Brat DJ, Brimijoin S. A paradigm for examing toxicant effects on variability, structure, and axonal transport of neurons in culture. Mol Neurobiol 6:125-136 (1992).
144. Brat DJ, Brimijoin S. Acrylamide and glycidamide impair neurite outgrowth in differentiating N1E 115 neuroblastoma without disturbing rapid bidirectional transport of organelles observed by video microscopy. J Neurochem 60:2145-2152 (1993).
145. Brat DJ, Windebank AJ, Brimijoin S. Emulsifier for intravenous cyclosporin inhibits neurite outgrowth, causes deficits in rapid axonal transport, and leads to structural abnormalities in differentiating N1E 115 neuroblastoma. J Pharmacol Exp Ther 261:803-810 (1992).
146. Harry GJ, Morell P, Bouldin TW. Acrylamide exposure preferentially impairs axonal transported glycoproteins in myelinated axons. J Neurosci Res 31:554-560 (1992).
147. Walum E, Peterson A. On the application of cultured neuroblastoma cells in chemical toxicity screening. J Toxicol Environ Health 13:511-520 (1984).
148. Bradbury MWB, Lightman SL. The blood brain interface. Eye 4:249-254 (1990).
149. Joo F. The blood brain barrier in vitro: ten years of research on microvessels isolated from brain. Neurochem Int 7:1-25 (1985).
150. Orlowski M, Sessa G, Green JP. ![]() -Glutymyl
transpeptidase in brain capillaries: possible site of a blood brain barrier
for amino acids. Science 184:66-68 (1974).
-Glutymyl
transpeptidase in brain capillaries: possible site of a blood brain barrier
for amino acids. Science 184:66-68 (1974).
151. Vorbrodt AW. Ultrastructural cytochemistry of blood brain barrier endothelia. Prog Histochem Cytochem 3:1-99 (1988).
152. Vannucci SJ. Developmental expression of GLUT1 and GLUT3 glucose transporter in rat brain. J Neurochem 62:1-5 (1994).
153. DeBault LE, Cancilla PA. Induction of ![]() -glutamyl
transpeptidase in isolated cerebral endothelial cells. Adv Exp Med Biol
131:79-88 (1980).
-glutamyl
transpeptidase in isolated cerebral endothelial cells. Adv Exp Med Biol
131:79-88 (1980).
154. Janzer RC, Raff MC. Astrocytes induce blood brain barrier properties in endothelial cells. Nature (London) 325:253-257 (1987).
155. Tontsch U, Bauer HC. Glial cells and neurons induce blood brain barrier related enzymes in cultured cerebral endothelial cells. Brain Res 539:247-253 (1991).
156. Walum E, Hansson E, Harvey AL. In vitro testing of neurotoxicity. ATLA 18:153-179 (1990).
157. Sellstrom A, Algers G, Karlsson B. Somanintoxication and the blood brain barrier. Fundam Appl Toxicol 5:S122-S126 (1985).
158. Kim YS, Lee MH, Wisniewski HM. Aluminum induced reversibly changes in permeability of the blood brain barrier to (14C) sucrose. Brain Res 377:286-291 (1986).
159. Brooks N, Kristt DA. Inhibition of amino acid transport and protein synthesis by mercury chloride and methylmercury in astrocytes: selectivity and reversibility. J Neurochem 53:1228-1237 (1989).
160. Ashani Y, Cartravas GN. Seizure-induced changes in the permeability of the blood-brain barrier following administration of anticholinesterase drugs to rats. Biochem Pharmacol 30(18):2593-2601 (1981).
161. Aschner M, and Aschner JL. Mercury neurotoxicity: mechanisms of blood-barin barrier transport. Neurosci Biobehav Rev 14:169-176 (1990).
162. Bressler JP, Goldstein. Mechanisms of lead neurotoxicity. Biochem Pharmacol 41:479-484 (1991).
163. Stanness KA, Guatteo E, Janigro D. A dynamic model of the blood-brain barrier "in vitro." Neurotoxicology 17:481-496 (1996).
164. Farber JL. The role of calcium in lethal cell injury. Chem Res Toxicol 3:503-508 (1990).
165. Miller RJ. The control of neuronal Ca2+-homeostasis. Prog Neurobiol 37:255-285 (1991).
166. Choi DW. Glutamate neurotoxicity in cortical cell culture is calcium dependent. Neurosci Lett 58:293-297 (1985).
167. Nicotera P, Bellomo G, Orrenium S. Calcium-mediated mechanisms in chemically induced cell death. Annu Rev Pharmacol Toxicol 32:449-470 (1992).
168. Verity MA. Ca2+-dependent processes as mediators of neurotoxicity. Neurotoxicology 13:139-148 (1992).
169. Zimmerman U-JP, Schlaepfer WW. Characterization of a brain calcium-activated protease that degrades neurofilament proteins. Biochemistry 21:3977-3983 (1982).
170. Drapeau P, Blaustein MP. Initial release of 3H-dopamine from rat striatal synaptosomes: correlation with calcium entry. J Neurosci 3:703-713 (1983).
171. Browning MD, Huganir R, Greengard P. Protein phosphorylation and neuronal function. J Neurochem 45:11-23 (1985).
172. Komulainen H, Bondy SC. Modulation of levels of free calcium within synaptosomes by organochlorine insecticides. J Pharmacol Exp Ther 241:575-581 (1987).
173. Komulainen H, Bondy SC. Increased free intrasynaptosomal Ca2+ by neurotoxic organometals: distinctive mechanisms. Toxicol Appl Pharmacol 88:77-86 (1987).
174. Komulainen H, Bondy SC. Increased free intracellular Ca2+ by toxic agents; an index of potential neurotoxicity? Trends Pharmacol Sci 9:154-156 (1988).
175. Verity MA, Sarafian TS, Guerra W, Ettinger A, Sharp J. Ionic modulation of triethyl lead neurotoxicity in cerebellar granule cell culture. Neurotoxicology 11:415-426 (1990).
176. Johnson JD, Conroy WG, Isom GE. Alteration of cytosolic calcium levels in PC12 cells by potassium cyanide. Toxicol Appl Pharmacol 88:217-224 (1987).
177. Doherty JD, Nishimura K, Kurihara N, Fujita T. Promotion of norepinephrine release and inhibition of calcium uptake by pyrethroids in rat brain synaptosomes. Pesticide Biochem Physiol 29:187-196 (1987).
178. Kodavanti PRS, Mundy WR, Tilson HA, Harry GJ. Effects of selected neuroactive chemicals on calcium transporting systems in rat cerebellum and on survival of cerebellar granule cells. Fundam Appl Toxicol 21:308-316 (1993).
179. Kodavanti PRS, Dae-Sup Shin, Tilson HA, Harry GJ. Comparative effects of two polychlorinated biphenyl congeners on calcium homeostasis in rat cerebellar granule cells. Toxicol Appl Pharmacol 123:97-106 (1993).
180. Koenig ML, Jope RS. Aluminum inhibits the fast phase of voltage-dependent calcium influx into synaptosomes. J Neurochem 49:316-320 (1987).
181. Wakui M, Itaya K, Birchall D, Petersen OH. Intracellular aluminum inhibits acetylcholine- and caffeine-evoked Ca2+ mobilization FEBS Lett 267:301-304 (1990).
182. Anghileri LJ. Effects of complexed iron and aluminum on brain calcium. Neurotoxicology 13:475-478 (1992).
183. Bottenstein JE. Differentiated properties of neuronal cell lines. In: Functionally Differentiated Cell Lines (Sato G, ed). New York:Alan R. Liss, 1981;154-184.
184. Schubert D, Carlisle W, Look C. Putative neurotransmitters in clonal cell lines. Nature 254:341-343 (1975).
185. Kimes B, Tarikas H, Schubert D. Neurotransmitter synthesis by two clonal nerve cell lines: changes with culture growth and morphological differentiation. Brain Res 79:291-295 (1974).
186. Greene l, Rein G. Synthesis, storage, and release of acetylcholine by noradrenergic pheochromocytoma cell line. Nature 268:349-351 (1977).
187. Reichardt LF, Patterson PH. Neurotransmitter synthesis and uptake by isolated sympathetic neurones in microcultures. Nature 270:147-151 (1977).
188. Johnson MI, Ross CD, Meyers M, Spitznagel EL, Bunge RP. Morphological and biochemical studies in the development of cholinergic properties in cultured sympathetic neurons. I: Correlative changes in choline acetyltransferase and synaptic vesicle cytochemistry. J Cell Biol 84:680-691 (1980).
189. Furshpan EJ, Potter DD, Landis SC. On the transmitter repertoire of sympathetic neurons in culture. Harvey Lect 76:149-191 (1981).
190. Higgins D, Lacovitti L, Joh TH, Burton H. The immunocytochemical localization of tyrosine hydroxylase within rat sympathetic neurons that release acetylcholine in culture. J Neurosci 1:126-131 (1981).
191. Heifi F, Hartikka J, Eckerstein F, Gnahn H, Heumann R, Schwab M. Nerve growth factor (NGF) increases choline acetyltransferase but not survival or fiber growth of cultured septal cholinergic neurons. Neuroscience 14:55-68 (1985).
192. Bunge R, Johnson M, Ross CD. Nature and nurture in development of the autonomic neuron. Science 199:1409-1416 (1978).
193. Bray D. Surface movements during growth of single explanted neurons. Proc Natl Acad Sci USA 65:905-910 (1970).
194. Chiappinelli V, Giacobini E, Pilar G, Uchimura H. Induction of cholinergic enzymes in chick ciliary ganglion and iris muscle cells during synapse formation. J Physiol 257:749-766 (1976).
195. Giacobini E, Pilar G, Suszkiw J, Uchimura H. Normal distribution and denervation changes of neurotransmitter related enzymes in cholinergic neurons. J Physiol 286:233-253 (1979).
196. Giacobini E. Aging of autonomic synapses. In: Advances in Cellular Neurobiology (Fedoroff S, Herz L, eds). New York:Academic Press, 1982;173-214.
197. Konig N, Wilkie MB, Lauder J. Dissection of monoaminergic neuronal groups from embryonic rat brain. In: A Dissection and Tissue Culture Manual of the Nervous System (Shahar A, de Vellis J, Vernadakis, Haber B, eds). New York:Alan R. Liss, 1989;26-29.
198. Whitaker-Azmitia PM, Azmitia EC. A tissue culture model of serotonin neurons for studying normal development and screening for drug teratogenicity. In: Alternative Methods in Toxicology and the Life Sciences. Vol 11: The World Congress on Alternatives and Animal Use in the Life Sciences: Education, Research, Testing (Goldberg A, van Zutphen LFM, eds). New York:Mary Ann Liebert, 1994;141-144.
199. Weissman AD, Johnson JE Jr. Assessing pyschoactive drug neurotoxicity in vitro: initial studies using NG108-15 neuroblastoma-glioma cells. In: In Vitro Toxicology: Mechanisms and New Technology. Vol 11: Alternatives Methods In Toxicology (Goldberg AM, ed). New York:Mary Ann Liebert, 1992;125-132.
200. Simerly RB, Swanson LW. The organization of neural inputs to the medial preoptic nucleus of the rat. J Comp Neurol 246:312-342 (1986).
201. Drewe JA, Childs GV, Kunze DL. Synaptic transmission between dissociated adult mammalian neurons and attached synaptic boutons. Science 241:1810-1813 (1988).
202. Bignami A, Dahl D, Rueger DC. Glial fibrillary acidic proteins in normal cells and in pathological conditions. Adv Cell Neurobiol 1:285-309 (1980).
203. Cookson MR, Pentreath VW. Alterations in the glial fibrillary acid protein content of primary astrocyte cultures for evaluation of glial cell toxicity. Toxicol In Vitro 8:351-359 (1994).
204. Cookson MR, Mead C, Austwick SM, Pentreath MW. Use of the MTT assay for estimating toxicity in primary astrocyte and C6 glioma cell cultures. Toxicol In Vitro 9:39-48 (1995).
205. Romero IA, Lister T, Richards MK, Sevillel MP, Wylie SP, Ray DE. Early metabolic changes during m-nitrobenzene neurotoxicity and the possible role of oxidative stress. Free Radic Biol Med 18:311-319 (1995).
206. Goldberg AM. An approach to the development of in vitro toxicological methods. Food Chem Toxicol 23:205-208 (1985).
207. Goldberg AM, ed. Alternataive Methods in Toxicology, in Vitro Toxicology Approaches to Validation, Vol 5. New York:Mary Ann Liebert, 1987.
208. Goldberg AM. Alternative Methods in Toxicology, Progress in in Vitro Toxicology, Vol 6. New York:Mary Ann Liebert, 1989.
209. Goldberg AM, Brookes N, Burt DR. The use of spinal cord cell cultures in the study of neurotoxicological agents. Toxicology 17:233-235 (1980).
210. Scala RA. Theoretical approaches to validation. In: In Vitro Toxicology Approaches to Validation. Alternative Methods in Toxicology, Vol 5 (Goldberg AM, ed). New York:Mary Ann Liebert, 1987;1-10.
211. Veronesi B. The use of cell culture for evaluating neurotoxicity. In: Neurotoxicology (Tilson H, Mitchell C, eds). New York:Raven Press, 1992;21-49.
212. Williams SP, Davenport-Jones J, Egan C, O'Hare S, Cookson M, McLean R, Garle MJ, Pentreath V, Atterwill CK. Phase I of an in vitro neurotoxicological prevalidation trial. Toxicol In Vitro 8:799-802 (1994).
Last Update: March 18, 1998