Toxicological
Defense Mechanisms and the Shape of Dose-Response Relationships
Environmental
Health Perspectives 106, Supplement 1, February 1998
1University of Texas Technical University, Lubbock, Texas; 2National Center for Toxicological Research, Jefferson, Arkansas
Key words: caloric restriction, dietary restriction, aging, calories, cancer, diet, carcinogenesis, glucocorticoids, life span, longevity, body mass index, adaptation, evolution
This paper is based on a presentation at The Third BELLE Conference on Toxicological Defense Mechanisms and the Shape of Dose-Response Relationships held 12-14 November 1996 in Research Triangle Park, NC. Manuscript received at EHP 29 May 1997; accepted 21 August 1997.The authors wish to thank A. Turturro for many thoughtful discussions. This work was sponsored in part by National Institute on Aging-National Center for Toxicological Research (NCTR) contract 224-86-0001 and the NCTR.
Address correspondence to Dr. J.E.A. Leakey, HFT-020, National Center for Toxicological Research, 3900 NCTR Road, Jefferson, AR 72079. Telephone: (501) 543-7916. Fax: (501) 543-7332. E-mail: jleakey@nctr.fda.gov
Abbreviations used: ACTH, adronocorticotrophic hormone; AVP, arginine vasopressin; BMI, body mass index; COX-2, cyclooxygenase isoform 2; CRF, corticotropin-releasing factor; CYP2C11, cytochrome P450 2C11; DHEA, dehydroepiandrosterone; EGF, epidermal growth factor; FSH, follicle-stimulating hormone; HPA, hypothalamo-pituitary-adrenal axis; HSP70, heat shock protein 70; IGF-1, insulinlike growth factor 1; LH, luteinizing hormone; NOSi, inducible form of nitric oxide synthase; PLAc, cytosolic phospholipase A2; PDGF, platelet-derived growth factor; TGF-ß, transforming growth factor beta.
The dose-response relationship between caloric intake and life span is not simple. In rodent models, for example, animals are at an increased risk of dying from protein-calorie malnutrition if dietary intake is extremely low. Interestingly, among survivors, caloric restriction is associated with a significant reduction in cancer incidence compared to that in ad libitum-fed controls. With moderate caloric restriction an optimal range may be found that generally supports reproduction, disease resistance, and longevity. The highest levels of caloric intake enhance growth and reproductive capacity but also significantly increase risk of morbidity, primarily because of development of specific cancers (7). These effects are reflected in the relationship between body weight and mortality, which is essentially J-shaped in rodent models (8,9) and the human population (10-13).
Under the usual experimental paradigms, calorically restricted animals receive a balanced reduction of the protein, carbohydrate, and fat content of the diet without a reduction of micronutrient content. Within a defined range of caloric restriction (typically 30-50% of ad libitum consumption), they show a decrease in the incidence and proliferative rate of spontaneous and chemically induced neoplasia. They also demonstrate a significant increase in maximum achievable life span compared to ad libitum-fed controls (6,14). Many biochemical and physiologic changes occur coincident with these effects but are difficult to distinguish from age-related pathologies (15). Therefore, for a mechanistic understanding, it is important to characterize and distinguish between the immediate adaptive response to reduced caloric intake and the resultant long-term effects on pathologic end points (7).
Nutrient stress such as fasting, starvation, or insulin-induced hypoglycemia results in elevated glucocorticoid levels, but unlike classic stress, hypothalamic release of corticotropin-releasing factor (CRF) does not appear to play a major role in initiating the glucocorticoid response (25-27). Rather, arginine vasopressin (AVP) plays the major role in the hypothalamus and the adrenal response to adrenocorticotropic hormone (ACTH) appears amplified by pancreatic polypeptide, which is secreted by the pancreas during periods of hypoglycemic stress (28,29). In addition, adrenal corticosterone secretion may be increased further by neural stimulation via the adrenal medulla (30,31). This results in elevated corticosterone concentrations in the absence of elevated ACTH (and by inference CRF) in both starved (25) and calorically restricted (27,32) rats. Under normal physiologic conditions, once the hypoglycemic crisis has been rectified, insulin levels will rise and, as suggested by in vitro experiments (33), may downregulate adrenal corticosterone secretion in favor of dehydroepiandrosterone (DHEA) secretion. Like insulin, DHEA is generally anabolic in function and reportedly antagonizes many of the effects of glucocorticoids (34-37).
However, during pathologic conditions such as Cushing's syndrome or prolonged excessive glucocorticoid therapy, natural feedback regulation is bypassed and a pathologic hyperglycemia develops, which is characterized by concurrent elevated insulin and glucocorticoid levels. Such conditions of hypercorticism concurrent with hyperinsulinemia, if prolonged, would be expected to result in pathologic conditions such as atherosclerosis and mature-onset diabetes (38). Classic stress appears to be controlled primarily by the hypothalamo-pituitary-adrenal axis (HPA). CRF and AVP secretion from the hypothalamus increase in response to interleukins or neuropeptides and stimulate ACTH secretion by the pituitary (39). Thus, plasma concentrations of both CRF and ACTH are increased in addition to serum corticosterone levels. CRF decreases hyperphagia (40) and is pyrogenic and an inflammatory mediator (41).
Apoptosis recently has been proposed to play an important role in inhibiting tumor development by eliminating damaged and genetically transformed cells from tumor-susceptible tissues (55-60). Apoptosis is characterized as differing from tissue necrosis in that only selected cells are eliminated and the resulting cell debris is immediately phagocytized by adjacent cells so that an inflammatory response is not initiated (61,62). Glucocorticoids induce apoptosis in lymphatic tissues (56) and fibroblasts (63) and possibly in mammary epithelium (61,64). Glucocorticoids may also selectively mediate the effects of transforming growth factor beta (TGF-ß) in stimulating apoptosis in preneoplastic hepatocytes (42,50).
Lipocortin 1 recently has been proposed to be a mediator of glucocorticoid-induced apoptosis. It is induced in apoptotic cells where it has been proposed to inhibit recognition of the dying cells by macrophages (61). Lipocortin 1 is also a substrate for transglutaminase, and covalently linked lipocortin dimers and polymers with other proteins may be produced in apoptopic cells (61,82,83). Recently, lipocortin 1 was shown to protect cultured rat thymocytes from H2O2-elicited necrosis. Glucocorticoid treatment, which induced lipocortin 1, stimulated apoptosis, whereas treatment with an antilipocortin 1 antibody enhanced necrosis (84).
Despite their global antiinflammatory effects, glucocorticoids have
been shown to potentiate certain aspects of the host defense system. For
example, they reportedly induce expression of both heat shock protein 70
(HSP70) (85) and the DNA repair enzyme O6-methylguarnine-DNA
methyltransferase (86) in certain tissues. They also potentiate the effects
of interleukin-6 and hepatocyte-stimulating factor in inducing hepatic
acute phase proteins such as Mn-superoxide dismutase and ![]() 2-macroglobulin
(87-91). Although both glucocorticoids and lymphocyte stimulatory agents
that are mediated via intracellular Ca2+ or protein kinase C
(e.g., calcium ionophors/phorbol esters, antibodies to the T-cell antigen
receptor) initiate apoptosis in maturing lymphocytes, they are mutually
antagonistic to the extent that glucocorticoids protect lymphocytes from
activation-induced apoptosis (92,93). Thus, the effects of glucocorticoids
on the inflammatory and immune systems are modulatory rather than simply
suppressive.
2-macroglobulin
(87-91). Although both glucocorticoids and lymphocyte stimulatory agents
that are mediated via intracellular Ca2+ or protein kinase C
(e.g., calcium ionophors/phorbol esters, antibodies to the T-cell antigen
receptor) initiate apoptosis in maturing lymphocytes, they are mutually
antagonistic to the extent that glucocorticoids protect lymphocytes from
activation-induced apoptosis (92,93). Thus, the effects of glucocorticoids
on the inflammatory and immune systems are modulatory rather than simply
suppressive.
Inflammation, necrosis, oxidative damage, and regenerative hyperplasia all play significant roles in chemically induced tumor promotion, and glucocorticoids have been shown to inhibit hyperplasia and neoplasia in a number of systems. For example, glucocorticoids are used therapeutically as antineoplastic agents in treating several types of leukemia and lymphoma (36,94) and suppress growth of certain lung or mammary adenocarcinomas (64,95-97). Dexamethasone inhibits both peroxisome-proliferator-induced and lead-nitrate-induced proliferative hyperplasia in rat liver (98,99). Glucocorticoids also induce connexin expression and stimulate gap junction formation in cultured hepatocytes and embryonic cells (100-102). Inflammatory agents such as phorbol esters promote, and glucocorticoids inhibit, papilloma formation in mouse skin (103).
Exposing adult rats to stress, hypercorticism, or glucocorticoid therapy reduces reproductive hormone levels in both sexes (7). In males, for example, glucocorticoids appear to inhibit LH-mediated testosterone synthesis by cultured rat Leydig cells (115) and dexamethasone treatment decreases (whereas adrenalectomy increases) serum testosterone levels in vivo (116,117). In females, glucocorticoids decrease follicle-stimulating hormone (FSH)-stimulated aromatase activity and estrogen production by ovarian granulosa cells (118), suppress ovulation, and inhibit ovarian prostaglandin metabolism (119). They also inhibit the preovulatory pituitary LH surge in female rats (120) and estradiol- and gonadotropin-releasing hormone-induced LH production in cultured rat pituitary cells (121). In male rats glucocorticoids inhibit pituitary secretion of prolactin (122) but not mean LH levels (123). However, CRF and stress inhibit pituitary LH secretion in both sexes (124,125).

Several factors differentiate the nutrient stress produced by caloric restriction from other stress situations or glucocorticoid therapy (7). First, unlike treatment with pharmacologic doses of synthetic glucocorticoids, hypercorticism resulting from nutrient stress involves the natural glucocorticoids corticosterone or cortisol. The effects of these natural glucocorticoids are mediated by serum transcortin and 11ß-hydroxysteroid dehydrogenase, which may protect tissues from extreme hypercorticism (7). Furthermore, unlike synthetic steroids such as dexamethasone, corticosterone and cortisol bind to both Type I and Type II glucocorticoid receptors, so the Type I receptor response is not inhibited concurrent with an excessive Type II receptor response (136).
Second, the hypercorticism exhibited by calorically restricted rodents differs from the continuously elevated serum corticosterone levels exhibited by starved or chronically stressed rodents in that corticosterone levels are increased above those of their ad libitum-fed counterparts only during a limited circadian period prior to and coincident with feeding activity (137). This type of intermittent hypercorticism appears less damaging to mitogenic processes than continuously elevated glucocorticoid levels (7).
Third, because the hypercorticism is a response to caloric deficit and potential hypoglycemia and occurs in conjunction with normal feedback regulatory systems, it is not associated with chronic hyperglycemia or hyperinsulinemia (16,127,138). Thus, insulin resistance and protein glycation, which are the usual pathologic consequences of glucocorticoid-induced hyperglycemia, should not occur. Instead, rates of intracellular glycation and oxidation of protein would be expected to decrease in peripheral tissues because of reduced glucose incorporation. Reduced collagen glycoxidation has been observed in skin from calorically restricted rats (139), and accumulative oxidative damage to both protein and DNA is reduced by caloric restriction in a number of tissues (43,140-142).
Fourth, under the usual conditions for caloric restriction experiments, significant hypercorticism only occurs during the early stages of restricted feeding (143,144). In most strains of rodent used in caloric restriction experiments, body weight gain is reduced in the restricted animals to an extent where the body weight difference between the restricted and ad libitum-fed animals equals or exceeds the caloric deficit (Figure 1). Thus, during the latter half of a calorically restricted rat's life span, its caloric consumption per gram body weight is equal to or greater than that of its ad libitum-fed counterpart. Under these conditions significant hypercorticism would not be required to protect the animal from potential hypoglycemia. As a consequence, during senescence, when rodents are most susceptible to tissue degeneration because of reduced capacity for cellular proliferation and reduced output of mitogenic hormones (131,146), serum corticosterone levels normally are no longer significantly increased in chronically calorically restricted animals (7,143,144).
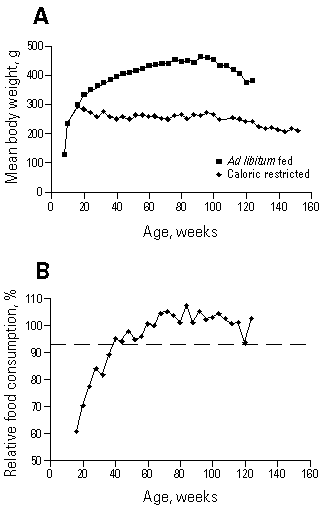
Figure 1. Influence of body weight on caloric consumption in calorically restricted Fischer 344 rats. Male F344 rats housed in a specific pathogen-free barrier facility at the National Center for Toxicological Research (Jefferson, AR) were placed on a vitamin-fortified NIH-31 diet at 40% of ad libitum food consumption, as described by Duffy et al. (147). (A) Body relative weight curves. (B) Relative food consumption (expressed as food consumed per gram body weight by the calorically restricted rats as a percentage of that consumed per gram body weight by the ad libitum-fed rats) as a function of age. By 50 weeks of age the calorically restricted rats consume equivalent amounts of food per gram body weight as their ad libitum-fed counterparts.
The effects of caloric restriction on biomarkers of mitogenesis are generally consistent with the occurrence of hypercorticism during the early but not the late stages of caloric restriction. For example, caloric restriction from 16 weeks of age abolishes growth hormone pulsatility in 6-month-old male Brown Norway rats, but pulsatility is restored in older animals (148). In male rats pulsatile growth hormone controls hepatic expression of both IGF-1 and sex-specific drug metabolizing enzymes such as cytochrome P450 2C11 (CYP2C11) (149,150). As expected from its effects on pulsatile growth hormone, caloric restriction decreases hepatic expression of both IGF-1 and CYP2C11 in young male rats (151,152). However, as the rats age hepatic IGF-1 and CYP2C11 expression decreases in the ad libitum-fed rats but is maintained by caloric restriction so that in old rats hepatic IGF-1 and CYP2C11 expression is greater in the calorically restricted animals (151,152). This age-dependent biphasic effect of caloric restriction is illustrated in Figure 2 and is a common feature of several of the reported effects of caloric restriction in rodents. These include cell proliferation rates in kidney, pancreas, and possibly liver from B6D2F1 mice (131), serum DHEA levels in Fischer 344 rats (153), and reproductive function in both rats and mice.
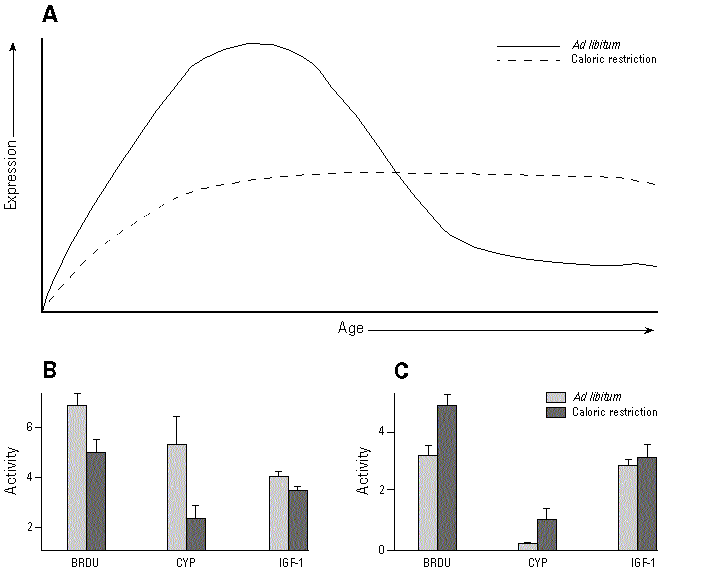
Figure 2. Age-dependent effects of caloric restriction. (A) Schematic representation of the effects of caloric restriction on mitogenic end points and reproductive function. Reduction in the early burst of activity delays the degradation of these systems in old age. Examples include cell proliferation in kidney tubule cells from B6D2F1 mice, as measured by in vivo labeling with BrdU (BRDU) (131); expression of hepatic CYP2C11 and its dependent activity, testosterone 16-hydroxylase in male F344 rats (CYP) (152); and expression of hepatic IGF-1 mRNA in male F344 rats (IGF-1) (151). Caloric restriction decreases these parameters (B) in young rodents but maintains them (C) in older rodents.
Effects on female reproductive funct-ion include delayed puberty (154,155), inhibition of LH pulsatility concurrent with hypercorticism (156), inhibition of ovulation (157), decreased litter size (158,159), increased lactational diestrus (160), and reduced milk production (161) during the initial period of caloric restriction and delayed reproductive senescence during the latter stage (135,158). In males the initial effects of caloric restriction include decreased LH pulsatility (162), reduced ratios of serum testosterone to estradiol (163), decreased sperm motility in rats (164,165), decreased prostrate weight, testicular sperm density, and fertility in mice (159). Long-term caloric restriction reduces testicular hyperplasia and delays Leydig cell adenoma formation in old male rats (163,166), whereas chronic feeding of a high-calorie diet reduced reproductive performance in old male CF-1 mice (167).
The antiinflammatory effects of caloric restriction are also generally consistent with effects resulting from hypercorticism. For example, caloric restriction reportedly induces lipocortin 1 immunoreactive proteins in rat liver (16), inhibits carrageenan-induced inflammation in mice (168), decreases 12-lipooxygenase activity in rat liver and testes (7), delays the onset of autoimmunity in autoimmune-prone mice (169), and inhibits promotion of mouse skin papillomas by phorbol esters (170,171). In the latter case adrenalectomy reversed the effect of caloric restriction. Interestingly, caloric restriction both potentiates regenerative hepatocyte proliferation in partially hepatectomized rats (172) and reduces cell proliferation while stimulating apoptosis in preneoplastic liver (173,174). Such an effect is consistent with the reported dual synergistic and antagonistic effects of glucocorticoids on TGF-ß in neoplastic and nonneoplastic hepatocytes (7,50).
Although older caloric-restricted mice exhibited improved cognitive function, motor performance, and reduced oxidative damage in the brain (132,133), caloric restriction did not inhibit hippocampal aging in rats, although it did not appear to be overtly detrimental to the hippocampus (109,175). However, dieting and dietary restriction reportedly impair cognitive function in humans (176). Despite potential endangerment to the hippocampus, moderate hypercorticism during nutrient stress would be expected to be beneficial because the alternative, hypoglycemia in conjunction with increased inflammatory activity, would pose a greater threat to the entire central nervous system.
Taken together, these effects suggest that caloric restriction in rodents produces a series of pleiotropic biochemical and physiologic effects consistent with a hypercorticism condition that is more severe in the early stages of caloric restriction than in the later stages and that occurs without concurrent hyperglycemia. The overall effect of this condition is to conserve energy by minimizing metabolism, proliferation, and nonessential functions in peripheral tissues. This in turn appears to minimize damage to the affected tissues so the progression of degenerative or neoplastic lesions is delayed.
The recent finding that the body weight of rodents used in cancer bioassays directly correlates with terminal incidence of background tumors (8,177) is also consistent with effects on growth and cell proliferation, which play a major role in mediating the antineoplastic effects of caloric restriction. These body weight-tumor correlations were demonstrated from analysis of the control animals from cancer bioassays conducted by the U.S. National Toxicology Program. In B6C3F1 mice terminal lung tumor incidence exhibited a positive correlation with body weight at 9 months on test, whereas terminal liver tumor incidence correlated optimally with body weight at 12 months on test (8,178). In Fischer 344 rats terminal pituitary tumor incidence exhibited a positive correlation with body weight at 13 months on test, whereas terminal leukemia incidence exhibited a positive correlation with body weight at 14 weeks (179). Interestingly, caloric restriction initiated at 6 weeks of age inhibited leukemia to a much greater extent than restriction initiated at 14 weeks, whereas pituitary adenoma formation was affected equally by both caloric restriction paradigms (179). This suggests that critical periods exist when rodents are most susceptible to subsequent development of specific cancer end points. This effect can also be demonstrated for background liver tumors in B6C3F1 mice (7).
It would appear, therefore, that the rate of growth during the early adult period of an organism's life determines its subsequent susceptibility to neoplastic or degenerative diseases, and rates of growth in part depend on glucocorticoid status and caloric intake. As stated above, glucocorticoids are a major component of the stress and inflammatory responses, where their primary functions appear to be: a) to globally reduce energy consumption so that energy is channeled to the site of trauma or inflammation, and b) to prevent excessive tissue damage due to overexpression of the inflammatory response. During severe nutrient stress hypercorticism allows an organism to conserve energy so that it may survive, but in the process growth and reproductive, immune, and cognitive functions may be compromised. However, caloric excess may be equally detrimental and result in overstimulated growth, uncontrolled cell proliferation, autoimmunity, inflammatory diseases, and neoplasia. Between these two extremes lies a physiologic window in which health and longevity are maximized (Figure 3). Hypercorticism as a hormonal response to nutrient stress appears common to most mammalian species and probably evolved as a mechanism to ensure survival of the species through periods of famine (180-183). In times of abundant food supply, rapid growth and fecundity are favored over endurance and longevity. Conversely, when food becomes scarce reproductive performance and growth are sacrificed in favor of extended total and reproductive life spans, thus increasing the probability that sufficient individuals will survive to restore the population when conditions improve.
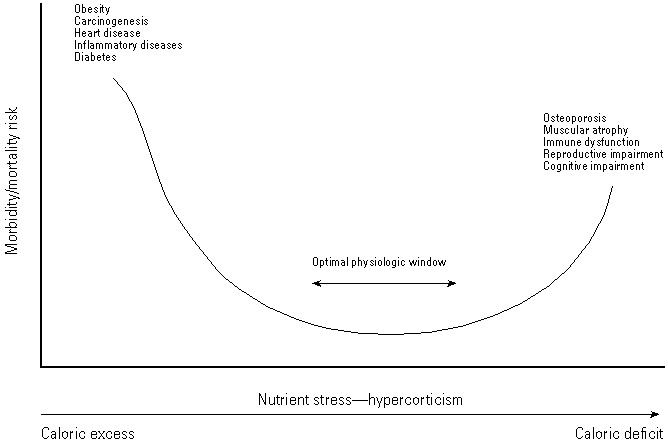
Figure 3. Relationship between caloric consumption and morbidity/mortality rates. Evidence from epidemiologicl and mechanistic studies suggests that the relationship between caloric consumption and morbidity/mortality is essentially J-shaped. High caloric consumption increases the risk of increased body growth, obesity, and associated pathologies, whereas extreme nutrient stress resulting from fasting, starvation, or excessive caloric restriction may increase the risk of tissue degeneration and associated pathologies.
Over the past 30 years the animal husbandry conditions used by commercial breeders and rodent-testing houses have been significantly modified to favor growth and fecundity (184,185). This has resulted in rodent strains that characteristically suffer from caloric excess and exhibit reduced life expectancy and many of the symptoms listed on the left side of Figure 3. When used in toxicity studies these animals are highly sensitive not only to chemically induced carcinogenesis but also to the acute effects of toxic chemicals (186). When used in moderation with these animals, dietary restriction decreases the incidence of both spontaneous and chemically induced carcinogenesis and also reportedly decreases the acute toxicity of several chemicals (186,187). Such effects are consistent with the antiinflammatory and antineoplastic effects of hypercorticism because a heightened inflammatory response amplifies the toxicity of carcinogens such as carbon tetrachloride (188-191). Although moderate dietary restriction is recommended for increasing life span and standardizing background tumor incidence between studies (192), excessive dietary restriction, whether a result of reduced food allocation or anorectic effects of the test chemical, should be avoided in cancer bioassays, as such conditions would render the bioassay insensitive for the detection of chemically induced tumors and reduce life expectancy because of the pathologic conditions listed on the right-hand side of Figure 3.
Although the relationship between body weight and morbidity is readily apparent in laboratory animals raised from similar genetic stock under controlled laboratory environments (177,178,197), establishing similar trends in the human population is considerably more difficult. It is now well established that genetic factors play a significant role in determining risk for both cancer and coronary heart disease (198,199) and behavioral factors such as cigarette smoking and heavy consumption of alcohol also influence susceptibility to these diseases (199-201). Nevertheless, when these factors are taken into consideration, dietary caloric consumption may be one of the most important risk factors for a spectrum of human degenerative diseases (4,202), and human epidemiology studies have established that increased body weight--or body mass index (BMI)--is positively correlated to a number of morbidity/mortality indices. These include overall mortality in both men and women (13,203); cardiovascular disease (10,13,204); breast, renal, and endometrial cancer in women (205-207); and colon cancer in men (208). As in animal studies (8) the relationship between BMI, weight, and morbidity/mortality risk is not always linear in human studies; J-curve profiles are frequently produced instead. Whereas some of the risk for low-body-weight individuals can be attributed to smoking (13), the studies quoted above used Western populations, which include relatively few individuals on low-calorie diets. Including populations from developing countries would be expected to produce mortality curves resembling that shown in Figure 3. Indeed it frequently has been noted that as caloric consumption relative to caloric demand increased in developed countries, disease susceptibilities changed from those characteristic of caloric deficit to those characteristic of caloric excess (209).
References
1. Pitot HC. The molecular determinants of carcinogenesis. Symp Fundam Cancer Res 39:187-196 (1986).
2. Pitot HC. Environmental modifiers in carcinogenesis. In: Host Factors in Human Carcinogensis. IARC Sci Publ No 39. Lyon:International Agency for Research on Cancer, 1982;165-176.
3. Doll R. An epidemiological perspective of the biology of cancer. Cancer Res 38:3573-3583 (1978).
4. Lutz WK, Schlatter J. Chemical carcinogens and overnutrition in diet-related cancer. Carcinogenesis 13:2211-2216 (1992).
5. Chow WH, Gridley G, McLaughlin JK, Mandel JS, Wacholder S, Blot WJ, Niwa S, Fraumeni JF Jr. Protein intake and risk of renal cell cancer. J Natl Cancer Inst 86:1131-1139 (1994).
6. Weindruch R, Walford R. Retardation of Aging and Disease by Dietary Restriction. Springfield, IL:Thomas Press, 1988.
7. Leakey JEA, Seng JE, Barnas CR, Baker VM, Hart RW. A mechanistic basis for the beneficial effects of dietary restriction on longevity and disease. Consequences for the interpretation of rodent toxicity studies. Int J Toxicol (in press).
8. Seilkop SK. The effect of body weight on tumor incidence and carcinogenicity testing in B6C3F1 mice and F344 rats. Fundam Appl Toxicol 24:247-259 (1995).
9. Hart RW, Turturro A. Is a new cancer risk assessment paradigm needed? BELLE Newslett 5:14-18 (1996).
10. Harris TB, Ballard-Barbasch R, Madans J, Makuc DM, Feldman JJ. Overweight, weight loss, and risk of coronary heart disease in older women. The NHANES I Epidemiologic Follow-up Study. Am J Epidemiol 137:1318-1327 (1993).
11. Peters ET, Seidell JC, Menotti A, Arayanis C, Dontas A, Fidanza F, Karvonen M, Nedeljkovic S, Nissinen A, Buzina R. Changes in body weight in relation to mortality in 6441 European middle-aged men: the Seven Countries Study. Int J Obesity Rel Metab Disorders 19:862-868 (1995).
12. Seidell JC, Verschuren WM, van Leer EM, Kromhout D. Overweight, underweight, and mortality. A prospective study of 48,287 men and women. Arch Intern Med 156:958-963 (1996).
13. Lee IM, Manson JE, Hennekens CH, Paffenbarger RS, Jr. Body weight and mortality. A 27-year follow-up of middle-aged men. JAMA 270:2823-2828 (1993).
14. Allaben WT, Chou MW, Pegram RA. Dietary restriction and toxicological endpoints: an historical overview. In: Biological Effects of Dietary Restriction (Fishbein L, ed). New York:Springer-Verlag, 1991;27-41.
15. Leakey JEA, Seng JE, Manjgaladze M, Kozlovskaya N, Xia S, Lee M-Y, Frame LT, Chen S, Rhodes CL, Duffy PH et al. Influence of caloric intake on drug metabolizing enzyme expression: relevance to tumorigenesis and toxicology testing. In: Dietary Restriction. Implications for the Design and Interpretation of Toxicity and Carcinogenicity Studies (Neumann DA, Hart RW, Robertson RT, eds). Washington:International Life Sciences Institute, 1995;167-180.
16. Leakey JEA, Chen S, Manjgaladze M, Turturro A, Duffy PH, Pipkin JL, Hart RW. Role of glucocorticoids and "caloric stress" in modulating the effects of caloric restriction in rodents. Ann NY Acad Sci 719:171-194 (1994).
17. Strack AM, Sebastian RJ, Schwartz MW, Dallman MF. Glucocorticoids and insulin: reciprocal signals for energy balance. Am J Physiol 268:R142-R149 (1995).
18. Dallman MF, Strack AM, Akana SF, Bradbury MJ, Hanson ES, Scribner KA, Smith M. Feast and famine: critical role of glucocorticoids with insulin in daily energy flow. Front Neuroendocrinol 14:303-347 (1993).
19. White BD, Martin RJ. Evidence for central mechanism of obesity in the Zucker rat: role of neuropeptide Y and leptin. Proc Soc Exp Biol Med 214:222-232 (1997).
20. Larsen PJ, Jessop DS, Chowdrey HS, Lightman SL, Mikkelsen JD. Chronic administration of glucocorticoids directly upregulates prepro-neuropeptide Y and Y1-receptor mRNA levels in the arcuate nucleus of the rat. J Neuroendocrinol 6:153-159 (1994).
21. Halaas JS, Gajiwala KS, Maffei M, Cohen SL, Chait BT, Rabinowitz D, Lallone RL, Burley SK, Freidman JM. Weight-reducing effects of the plasma protein encoded by the obese gene. Nature 269:543-546 (1995).
22. MacDougald OA, Hwang C-S, Fan H, Lane MD. Regulated expression of the obese gene product (leptin) in white adipose tissue and 3T3-L1 adiposites. Proc Natl Acad Sci USA 92:9034-9037 (1995).
23. Pelleymounter MA, Cullen MJ, Baker MB, Hecht R, Winters D, Boone T, Collins F. Effects of the obese gene product on body weight regulation in ob/ob mice. Nature 269:540-543 (1995).
24. Saladin R, De Vos P, Guerre-Millo M, Lecturque A, Girard J, Staels B, Auwerx J. Transient increase in obese gene expression after food intake or insulin administration. Nature 377:527-529 (1995).
25. Suemaru S, Hashimoto K, Hattori T, Inoue H, Kageyama J, Ota Z. Starvation-induced changes in rat brain corticotropin-releasing factor (CRF) and pituitary-adrenocortical response. Life Sci 39:1161-1166 (1986).
26. Muret L, Priou A, Oliver C, Grino M. Stimulation of adrenocorticotropin secretion by insulin-induced hypoglycemia in the developing rat involves arginine vasopressin but not corticotropin-releasing factor. Endocrinology 130:2725-2732 (1992).
27. Leal AM, Forsling ML, Moreira AC. Diurnal variation of the pituitary-adrenal and AVP responses to stress in rats under food restriction. Life Sci 56:191-198 (1995).
28. Andreis PG, Tortorella C, Nussdorfer GG. Pancreatic polypeptide stimulates corticosterone secretion by isolated rat adrenocortical cells. Life Sci 53:1353-1356 (1993).
29. Mazzocchi G, Malendowicz LK, Gottardo G, Meneghelli V, Nussdorfer GG. Pancreatic polypeptide enhances plasma glucocorticoid concentration in rats: possible role in hypoglycemic stress. Life Sci 56:595-600 (1995).
30. Andreis PG, Neri G, Nussdorfer GG. Corticotropin-releasing hormone (CRH) directly stimulates corticosterone secretion by the rat adrenal gland. Endocrinology 128:1198-1200 (1991).
31. Tsujimoto S, Okumura Y, Kamei C, Tasaka K. Effects of intracerebroventricular injection of histamine and related compounds on corticosterone release in rats. Br J Pharmacol 109:807-813 (1993).
32. Han E-S, Levin N, Bengani N, Roberts JL, Suh Y, Karelus K, Nelson JF. Hyperadrenocorticism and food restriction-induced life extension in the rat: evidence for divergent regulation of pituitary proopiomelanocortin RNA and adrenocorticotropic hormone biosynthesis. J Gerontol 50A:B288-B294 (1995).
33. Kramer RE, Buster JE, Andersen RN. Differential modulation of ACTH-stimulated cortisol and androstenedione secretion by insulin. J Steroid Biochem 36:33-42 (1990).
34. Shafagoj Y, Opoku J, Qureshi D, Regelson W, Kalimi M. Dehydroepiandrosterone prevents dexamethasone-induced hypertension in rats. Am J Physiol 263:E210-E213 (1992).
35. Kalimi M, Shafagoj Y, Loria R, Padgett D, Regelson W. Anti-glucocorticoid effects of dehydroepiandrosterone (DHEA). Mol Cell Biochem 131:99-104 (1994).
36. Regelson W, Kalimi M. Dehydroepiandrosterone (DHEA)--the multifunctional steroid. II: Effects on the CNS, cell proliferation, metabolic and vascular, clinical and other effects. Mechanism of action? Ann NY Acad Sci 719:564-575 (1994).
37. Regelson W, Loria R, Kalimi M. Dehydroepiandrosterone (DHEA)--the "mother steroid." I: Immunologic action. Ann NY Acad Sci 719:553-563 (1994).
38. Landfield PW. Modulation of brain aging correlates by long-term alterations of adrenal steroids and neurally-active peptides. Prog Brain Res 72:279-300 (1987).
39. Buckingham JC, Loxley HD, Taylor AD, Flower RJ. Cytokines, glucocorticoids and neuroendocrine function. Pharmacol Res 30:35-42 (1994).
40. Debons AF, Zurek LD, Tse CS, Abrahamsan S. Central nervous system control of hyperphagia in hypothalamic obesity: dependence on adrenal glucocorticoids. Endocrinology 118:1678-1681 (1986).
41. Strijbos PJ, Hardwick AJ, Relton JK, Carey F, Rothwell NJ. Inhibition of central actions of cytokines on fever and thermogenesis by lipocortin-1 involves CRF. Am J Physiol 263:E632-E636 (1992).
42. Feuers RJ, Weindruch R, Hart RW. Caloric restriction, aging and antioxidant enzymes. Mutat Res 295:191-200 (1993).
43. Wachsman JT. The beneficial effects of dietary restriction: reduced oxidative damage and enhanced apoptosis. Mutat Res 350:25-34 (1996).
44. Mehls O, Tönshoff B, Kovàcs G, Mayer C, Schurek J, Oh J. Interaction between glucocorticoids and growth hormone. Acta Paediatr 82:77-82 (1993).
45. Suh DY, Hunt TK, Spencer EM. Insulin-like growth factor-I reverses the impairment of wound healing induced by corticosteroids in rats. Endocrinology 131:2399-2403 (1992).
46. Wehrenberg WB, Janowski BA, Piering AW, Culler F, Jones KL. Glucocorticoids: potent inhibitors and stimulators of growth hormone secretion. Endocrinology 126:3200-3203 (1990).
47. Planas B, Kolb PE, Raskind MA, Miller MA. Galanin-binding sites in the female rat brain are regulated across puberty yet similar to the male pattern in adulthood. Neuroendocrinology 61:646-654 (1995).
48. Luo J, Murphy LJ. Dexamethasone inhibits growth hormone induction of insulin-like growth factor-1 (IGF-1) messenger ribonucleic acid (mRNA) in hypophysectomized rats and reduces IGF-1 mRNA abundance in the intact rat. Endocrinology 125:165-171 (1989).
49. Kim SB, Yang WS, Lee OS, Lee KP, Park JS, Na DS. Lipocortin-1 inhibits proliferation of cultured human mesangial cells. Nephron 74:39-44 (1996).
50. Wollenberg GK, LaMarre J, Semple E, Farber E, Gauldie J, Hayes MA. Counteracting effects of dexamethasone and *2-macroglobulin on inhibition of proliferation of normal and neoplastic rat hepatocytes by transforming growth factors-ß type 1 and type 2. Int J Cancer 47:311-316 (1991).
51. Cooke BA, Platts EA, Abayasekera R, Kurlak LO, Schulster D, Sullivan MHF. Control of multiple transducing systems by LH which results in the modulation of adenylate cyclase, protein kinase C, lipoxygenases and cyclooxygenases. J Reprod Fert Suppl 37:139-145 (1989).
52. Cooke BA, Dirami G, Chaudry L, Choi MKS, Abayasekera DRE, Phipp L. Release of arachidonic acid and the effects of corticosteroids on steroidogenesis in rat testes Leydig cells. J Steroid Biochem Molec Biol 40:465-471 (1991).
53. Czerwinski SM, Kurowski TT, McKee EE, Zak R, Hickson RC. Myosin heavy chain turnover during cardiac mass changes by glucocorticoids. J Appl Physiol 70:300-305 (1991).
54. Haddad F, Bodell PW, McCue SA, Herrick RE, Baldwin KM. Food restriction-induced transformations in cardiac functional and biochemical properties in rats. J Appl Physiol 74:606-612 (1993).
55. Canman CE, Chen CY, Lee MH, Kastan MB. DNA damage responses: p53 induction, cell cycle perturbations, and apoptosis. Cold Spring Harbor Symp Quant Biol 59:277-286 (1994).
56. Schwartzman RA, Cidlowski JA. Glucocorticoid-induced apoptosis of lymphoid cells. Int Arch Allergy Immunol 105:347-354 (1994).
57. Benedetti A, Di Sario A, Svegliati Baroni G, Jezequel AM. Transforming growth factor ß1 increases the number of apoptotic bodies and decreases intracellular pH in isolated periportal and perivenular rat hepatocytes. Hepatology 22:1488-1498 (1995).
58. Gantner F, Leist M, Jilg S, Germann PG, Freudenberg MA, Tiegs G. Tumor necrosis factor-induced hepatic DNA fragmentation as an early marker of T cell-dependent liver injury in mice. Gastroenterology 109:166-176 (1995).
59. Lee JM, Bernstein A. Apoptosis, cancer and the p53 tumour suppressor gene. Cancer Metastasis Rev 14:149-161 (1995).
60. Witty JP, Lempka T, Coffey RJ Jr, Matrisian LM. Decreased tumor formation in 7,12-dimethylbenzanthracene-treated stromelysin-1 transgenic mice is associated with alterations in mammary epithelial cell apoptosis. Cancer Res 55:1401-1406 (1995).
61. Mckanna JA. Lipocortin 1 in apoptosis: mammary regression. Anat Rec 242:1-10 (1995).
62. Columbano A. Cell death: current difficulties in discriminating apoptosis from necrosis in the context of pathological processes in vivo. J Cell Biochem 58:181-190 (1995).
63. O'Banion MK, Levenson RM, Brinckmann UG, Young DA. Glucocorticoid modulation of transformed cell proliferation is oncogene specific and correlates with effects on c-myc levels. Mol Endocrinol 6:1371-1380 (1992).
64. Alexander DB, Goya L, Webster MK, Haraguchi T, Firestone GL. Glucocorticoids coordinately disrupt a transforming growth factor * autocrine loop and suppress the growth of 13762NF-derived Con8 rat mammary adenocarcinoma cells. Cancer Res 53:1808-1815 (1993).
65. Flower RL. Lipocortin and the mechanism of action of the glucoorticoids. Br J Pharmacol 94:987-1015 (1988).
66. Selye H. The general adaptation syndrome and the diseases of adaptation. J Clin Endocrinol Metab 6:117-126 (1946).
67. Munck A, Guyre PM, Holbrook NJ. Physiological functions of glucocorticoids in stress and their relation to pharmacological actions. Endocrinol Rev 5:25-44 (1984).
68. Flower RJ, Rothwell NJ. Lipocortin-1: cellular mechanisms and clinical relevance. Trends Pharmacol Sci 15:71-76 (1994).
69. Croxtall JD, Choudhury Q, Tokumoto H, Flower RJ. Lipocortin-1 and the control of arachidonic acid release in cell signalling. Glucocorticoids inhibit G protein-dependent activation of cPLA2 activity. Biochem Pharmacol 50:465-474 (1995).
70. Croxtall JD, Choudhury Q, Newman S, Flower RJ. Lipocortin 1 and the control of cPLA2 activity in A549 cells. Glucocorticoids block EGF stimulation of cPLA2 phosphorylation. Biochem Pharmacol 52:351-356 (1996).
71. Loxley HD, Cowell AM, Flower RJ, Buckingham JC. Modulation of the hypothalamo-pituitary-adrenocortical responses to cytokines in the rat by lipocortin 1 and glucocorticoids: a role for lipocortin 1 in the feedback inhibition of CRF-41 release? Neuroendocrinology 57:801-814 (1993).
72. Wang W, Creutz CE. Role of the amino-terminal domain in regulating interactions of annexin I with membranes: effects of amino-terminal truncation and mutagenesis of the phosphorylation sites. Biochemistry 33:275-282 (1994).
73. Masferrer JL, Steibert K, Zweifel B, Needleman P. Endogenous glucocorticoids regulate an inducible cyclooxygenase enzyme. Proc Natl Acad Sci USA 89:3917-3921 (1992).
74. Masferrer JL, Zweifel BS, Manning PT, Hauser SD, Leahy KM, Smith WG, Isakson PC, Seibert K. Selective inhibition of inducible cyclooxygenase 2 in vivo is antiinflammatory and nonulcerogenic. Proc Natl Acad Sci USA 91:3228-3232 (1994).
75. Seibert K, Masferrer JL. Role of inducible cyclooxygenase (COX-2) in inflammation. Receptor 4:17-23 (1994).
76. Salvemini D, Manning PT, Zweifel BS, Seibert K, Connor J, Currie MG, Needleman P, Masferrer JL. Dual inhibition of nitric oxide and prostaglandin production contributes to the antiinflammatory properties of nitric oxide synthase inhibitors. J Clin Invest 96:301-308 (1995).
77. Schmidt HHHW, Lohmann SM, Walter U. The nitric oxide and cGMP signal transduction system: regulation and mechanism of action. Biochim Biophys Acta 1178:153-175 (1993).
78. Geller DA, de Vera ME, Russell DA, Shapiro RA, Nussler AK, Simmons RL, Billiar TR. A central role for IL-1ß in the in vitro and in vivo regulation of hepatic inducible nitric oxide synthase. IL-1ß induces hepatic nitric oxide synthesis. J Immunol 155:4890-4898 (1995).
79. Lilja I, Dimberg J, Sjodahl R, Tagesson C, Gustafson-Svard C. Effects of endotoxin and dexamethasone on group I and II phospholipase A2 in rat ileum and stomach. Gut 35:40-45 (1994).
80. Wu CC, Croxtall JD, Perretti M, Bryant CE, Thiemermann C, Flower RJ, Vane JR. Lipocortin 1 mediates the inhibition by dexamethasone of the induction by endotoxin of nitric oxide synthase in the rat. Proc Natl Acad Sci USA 92:3473-3477 (1995).
81. Newman SP, Flower RJ, Croxtall JD. Dexamethasone suppression of IL-1 beta-induced cyclooxygenase 2 expression is not mediated by lipocortin-1 in A549 cells. Biochem Biophys Res Commun 202:931-939 (1994).
82. Pepinsky RB, Sinclair LK, Chow EP, O'Brine Greco B. Dimeric form of lipocortin-1 in human placenta. Biochem J 263:97-103 (1989).
83. Ando Y, Imamura S, Owada MK, Kannagi R. Calcium-induced intracellular cross-linking of lipocortin I by tissue transaminase in A431 cells. J Biol Chem 266:1101-1108 (1991).
84. Sakamoto T, Repasky W, Uchida K, Hirata A, Hirata F. Modulation of cell death pathways to apoptosis and necrosis of H2O2-treated rat thymocytes by lipocortin-1. Biochem Biophys Res Commun 220:643-647 (1996).
85. Heufelder AE, Wenzel BE, Bahn RS. Glucocorticoids modulate the synthesis and expression of a 72 kDa heat shock protein in cultured Graves' retroocular fibroblasts. Acta Endocrinol 128:41-50 (1993).
86. Grombacher T, Mitra S, Kaina B. Induction of the alkyltransferase (MGMT) gene by DNA damaging agents and the glucocorticoid dexamethasone and comparison with the response of base excision repair genes. Carcinogenesis 17:2329-2336 (1996).
87. Bauer J, Tran-Thi TA, Northoff H, Hirsch F, Schlayer HJ, Gerok W, Heinrich PC. The acute-phase induction of *2-macroglobulin in rat hepatocyte primary cultures: action of a hepatocyte-stimulating factor, triiodothyronine and dexamethasone. Eur J Cell Biol 40:86-93 (1986).
88. Kurokawa S, Ishibashi H, Shirahama M, Hayashida K, Tsuchiya Y, Sakaki Y, Niho Y. Production of hepatocyte stimulating factor of rat Kupffer cells induced by lipopolysaccharide: partial characterization and effects on *2 macroglobulin gene expression in cultured adult rat hepatocytes. J Clin Lab Immunol 25:131-137 (1988).
89. Marinkovic S, Jahreis GP, Wong GG, Baumann H. IL-6 modulates the synthesis of a specific set of acute phase plasma proteins in vivo. J Immunol 142:808-812 (1989).
90. Dougall WC, Nick HS. Manganese superoxide dismutase: a hepatic acute phase protein regulated by interleukin-6 and glucocorticoids. Endocrinology 129:2376-2384 (1991).
91. Hocke GM, Barry D, Fey GH. Synergistic action of interleukin-6 and glucocorticoids is mediated by the interleukin-6 response element of the rat *2 macroglobulin gene. Mol Cell Biol 12:2282-2294 (1992).
92. Garcia-Leme J, Fortes ZB, Sannomiya P, Farsky SP. Insulin, glucocorticoids and the control of inflammatory responses. Agents Actions 36:99-118 (1992).
93. Zacharchuk CM, Mercerp M, Chakaborti PK, Simons SS Jr, Ashwell JD. Programmed T lymphocyte death. Cell-activated and steroid-induced pathways are mutually antagonistic. J Immunol 145:4037-4045 (1990).
94. King LB, Ashwell JD. Thymocyte and T cell apoptosis: is all death created equal? Thymus 23:209-230 (1994).
95. Croxtall JD, Flower RJ. Lipocortin 1 mediates dexamethasone-induced growth arrest of the A549 lung adenocarcinoma cell line. Proc Natl Acad Sci USA 89:3571-3575 (1992).
96. Wattenberg LW, Estensen RD. Chemopreventive effects of myo-inositol and dexamethasone on benzo[a]pyrene and 4-(methylnitrosoamino)-1-(3-pyridyl)-1-butanone-induced pulmonary carcinogenesis in female A/J mice. Cancer Res 56:5132-5135 (1996).
97. Hofmann J, Kaiser U, Maasberg M, Havemann K. Glucocorticoid receptors and growth inhibitory effects of dexamethasone in human lung cancer cell lines. Eur J Cancer 31A:2053-2058 (1995).
98. Duncan GS, Peers SH, Carey F, Forder R, Flower RJ. Dexamethasone inhibits induction of liver tumor necrosis factor-* mRNA and liver growth induced by lead nitrate and ethylene dibromide. Am J Pathol 145:951-958 (1993).
99. Rao MS, Subbarao V. Effect of dexamethasone on ciprofibrate-induced cell proliferation and peroxisome proliferation. Fundam Appl Toxicol 35:78-83 (1997).
100. Roseng LE, Rivedal E. Effect of glucocorticoids on TPA-induced inhibition of gap-junctional communication and morphological transformation in Syrian hamster embryo cells. Cancer Lett 72:25-30 (1993).
101. Kwiatkowski AP, Baker TK, Klaunig JE. Comparison of glucocorticoid-mediated changes in the expression and function of rat hepatocyte gap junctional proteins. Carcinogenesis 15:1753-1757 (1994).
102. Ren P, de Feijter AW, Paul DL, Ruch RJ. Enhancement of liver cell gap junction protein expression by glucocorticoids. Carcinogenesis 15:1807-1813 (1994).
103. Slaga TJ, DiGiovanni J, Winberg LD, Budunova IV. Skin carcinogenesis: characteristics, mechanisms, and prevention. Prog Clin Biol Res 391:1-20 (1995).
104. Gaynon PS, Lustig RH. The use of glucocorticoids in acute lymphoblastic leukemia of childhood. Molecular, cellular, and clinical considerations. J Pediat Hematol Oncol 17:1-12 (1995).
105. McEwen BS, Sapolsky RM. Stress and cognitive function. Curr Opin Neurobiol 5:205-216 (1995).
106. Rothwell NJ, Relton JK. Involvement of cytokines in acute neurodegeneration in the CNS. Neurosci Biobehav Rev 17:217-227 (1993).
107. Sapolsky R, Krey L, McEwen B. The neuroendocrinology of stress and aging: the glucocorticoid cascade hypothesis. Endocrinol Rev 7:284-302 (1993).
108. Stein-Behrens BA, Sapolsky RM. Stress, glucocorticoids and aging. Aging Clin Exp Res 4:197-210 (1992).
109. Landfield PW, Eldridge JC. Evolving aspects of the glucocorticoid hypothesis of brain aging: hormonal modulation of neuronal calcium homeostasis. Neurobiol Aging 15:579-588 (1994).
110. Starkman MN, Schteingart DE, Schork MA. Cushing's syndrome after treatment: changes in cortisol and ACTH levels, and amelioration of the depressive syndrome. Psych Res 19:177-188 (1986).
111. Newcomer JW, Craft S, Hershey T, Askins K, Bardgett ME. Glucocorticoid-induced impairment in declarative memory performance in adult humans. J Neurosci 14:2047-2053 (1994).
112. Breitner JCS, Gau BA, Welsh KA, Plassman BL, McDonald WM, Helms MJ, Amthony JC. Inverse association of anti-inflammatory treatments and Alzheimer's disease: initial results of a co-twin control study. Neurology 44:227-232 (1994).
113. Black MD, Carey F, Crossman AR, Relton JK, Rothwell NJ. Lipocortin-1 inhibits NMDA-reception-mediated neuronal damage in the striatum of the rat. Brain Res 585:135-140 (1992).
114. Rothwell NJ, Relton JK. Involvement of interleukin-1 and lipocortin-1 in ischaemic brain damage. Cerebrovasc Brain Metab Rev 5:178-198 (1993).
115. Welsh THJ, Bambino TH, Hsueh AJW. Mechanism of glucocorticoid-induced suppression of testicular androgen biosynthesis in vitro. Biol Reprod 27:1138-1146 (1982).
116. Bambino TH, Hsueh AJW. Direct inhibitory effect of glucocorticoids upon testicular luteinizing hormone receptor and steroidogenesis in vivo and in vitro. Endocrinology 108:2142-2147 (1981).
117. Gao HB, Shan LX, Monder C, Hardy MP. Suppression of endogenous corticosterone levels in vivo increases the steroidogenic capacity of purified rat Leydig cells in vitro. Endocrinology 137:1714-1718 (1996).
118. Hsueh AJW, Erickson GF. Glucocorticoid inhibition of FSH-induced estrogen production in cultured rat granulosa cells. Steroids 32:639-648 (1978).
119. Inazu N, Iwata N, Satoh T. Inhibitory effect of glucocorticoid and stimulatory effect of human chorionic gonadotropin on ovarian carbonyl reductase in rats. Life Sci 46:841-848 (1990).
120. Baldwin DM, Sawyer CH. Effects of dexamethasone on LH release and ovulation in the cyclic rat. Endocrinology 94:1397-1403 (1974).
121. Kamel F, Kubajak CL. Modulation of gonadotropin secretion by corticosterone: interaction with gonadal steroids and mechanism of action. Endocrinology 121:561-568 (1987).
122. Lopez-Calderon A, Ariznavarreta C, Calderon MD, Tresguerres JA, Gonzalez-Quijano MI. Role of the adrenal cortex in chronic stress-induced inhibition of prolactin secretion in male rats. J Endocrinol 120:269-273 (1989).
123. Lopez-Calderon A, Ariznavarreta C, Calderon MD, Tresguerres JA. Gonadotropin inhibition during chronic stress: role of the adrenal gland. J Steroid Biochem 27:609-614 (1987).
124. Rivier C, Vale W. Influence of corticotropin-releasing factor on reproductive functions in the rat. Endocrinology 114:914-921 (1984).
125. Rivier C, Vale W. Effect of long-term administration of corticotropin-releasing factor on the pituitary-adrenal and pituitary gonadal axis in the male rat. J Clin Invest 75:689-694 (1985).
126. Kalant N, Stewart J, Kaplan R. Effect of diet restriction on glucose metabolism and insulin responsiveness in aging rats. Mech Ageing Devel 46:89-104 (1988).
127. Harris SB, Gunion MW, Rosenthal MJ, Walford RL. Serum glucose, glucose tolerance, corticosterone and free fatty acids during aging in energy restricted mice. Mech Ageing Devel 73:209-221 (1994).
128. Bodkin NL, Ortmeyer HK, Hansen BC. Long-term dietary restriction in older-aged rhesus monkeys: effects on insulin resistance. J Gerontol 50:B142-B147 (1995).
129. Ohneda M, Inman LR, Unger RH. Caloric restriction in obese pre-diabetic rats prevents beta-cell depletion, loss of beta-cell GLUT 2 and glucose incompetence. Diabetologia 38:173-179 (1995).
130. Lewis SE, Goldspink DF, Phillips JG, Merry BJ, Holehan AM. The effects of aging and chronic dietary restriction on whole body growth and protein turnover in the rat. Exp Gerontol 20:253-263 (1985).
131. Wolf NS, Penn PE, Jiang D, Fei RG, Pendergrass WR. Caloric restriction: conservation of in vivo cellular replicative capacity accompanies life-span extension in mice. Exp Cell Res 217:317-323 (1995).
132. Forster MJ, Dubey A, Dawson KM, Stutts WA, Lal H, Sohal RS. Age-related losses of cognitive function and motor skills in mice are associated with oxidative protein damage in the brain. Proc Natl Acad Sci USA 93:4765-4769 (1996).
133. Lal H, Forster MJ, Sohal RS. Oxidative brain damage in aged mice. Protection by caloric reduction [Abstract]. Ann NY Acad Sci 765:308 (1995).
134. Holehan AM, Merry BJ. Modification of the oestrous cycle hormonal profile by dietary restriction. Mech Ageing Dev 32:63-76 (1985).
135. Keenan KP, Soper KA, Hertzog PR, Gumprecht LA, Smith PF, Mattson BA, Ballam GC, Clark RL. Diet, overfeeding, and moderate dietary restriction in control Sprague-Dawley rats. II: Effects on age-related proliferative and degenerative lesions. Toxicol Pathol 23:287-302 (1995).
136. Reul JM, Van den Bosch FR, De Kloet ER. Differential response of type I and type II corticosteroid receptors to changes in plasma steroid level and circadian rhythmicity. Neuroendocrinology 45:407-412 (1987).
137. Ma Q, Dannan GA, Guengerich FP, Yang CS. Similarities and differences in the regulation of hepatic cytochrome P-450 enzymes by diabetes and fasting in male rats. Biochem Pharmacol 38:3179-3184 (1989).
138. Masoro EJ, Shimokawa I, Yu BP. Retardation of the aging processes in rats by food restriction. Ann NY Acad Sci 621:337-352 (1991).
139. Sell DR, Lane MA, Johnson WA, Masoro EJ, Mock OB, Reiser KM, Fogarty JF, Cutler RG, Ingram DK, Roth GS et al. Longevity and the genetic determination of collagen glycoxidation kinetics in mammalian senescence. Proc Natl Acad Sci USA 93:485-490 (1996).
140. Sohal RS, Agarwal S, Candas M, Forster MJ, Lal H. Effect of age and caloric restriction on DNA oxidative damage in different tissues of C57BL/6 mice. Mech Ageing Dev 76:215-224 (1994).
141. Djuric Z, Lu MH, Lewis SM, Luongo DA, Chen XW, Heilbrun LK, Reading BA, Duffy PH, Hart RW. Oxidative damage levels in rats fed low-fat, high-fat, or calorie-restricted diets. Toxicol Appl Pharmacol 115:156-160 (1992).
142. Sohal RS, Ku HH, Agarwal S, Forster MJ, Lal H. Oxidative damage, mitochondrial oxidant generation and antioxidant defenses during aging and in response to food restriction in the mouse. Mech Ageing Dev 74:121-133 (1994).
143. Stewart J, Meaney MJ, Aitken D, Jensen L, Kalant N. The effects of acute and life-long food restriction on basial and stress-induced serum corticosterone levels in young and aged rats. Endocrinology 123:1934-1941 (1988).
144. Sabatino F, Masoro EJ, McMahan CA, Kuhn RW. Assessment of the role of the glucocorticoid system in aging processes and in the action of the food restriction. J Gerontol 46:B171-B179 (1991).
145. Hart RW, Leakey JE, Chou M, Duffy PH, Allaben WT, Feuers RJ. Modulation of chemical toxicity by modification of caloric intake. Adv Exp Med Biol 322:73-81 (1992).
146. Pendergrass WR, Li Y, Jiang D, Fei RG, Wolf NS. Caloric restriction: conservation of cellular replicative capacity in vitro accompanies life-span extension in mice. Exp Cell Res 217:309-316 (1995).
147. Duffy PH, Feuers RJ, Leakey JEA, Nakamura KD, Turturro A, Hart RW. Effect of chronic caloric restriction on the physiological variables related to energy metabolism in the Fischer 344 rat. Mech Ageing Dev 48:117-133 (1989).
148. Sonntag WE, Xu X, Ingram RL, D'Costa A. Moderate caloric restriction alters the subcellular distribution of somatostatin mRNA and increases growth hormone pulse amplitude in aged animals. Neuroendocrinology 61:601-608 (1995).
149. Hynes MA, Van Wyk JJ, Brooks PJ, D'Ercole AJ, Jansen M, Lund PK. Growth hormone dependence of somatomedin-C/insulin-like growth factor-I and insulin-like growth factor-II messenger ribonucleic acids. Mol Endocrinol 1:233-242 (1987).
150. Westin S, Tollet P, Strom A, Mode A, Gustafsson JA. The role and mechanism of growth hormone in the regulation of sexually dimorphic-P450 enzymes in rat liver. J Steroid Biochem Mol Biol 43:1045-1053 (1992).
151. Breese CR, Ingram RL, Sonntag W. Influence of age and long-term dietary restriction on plasma insulin-like growth factor-1 (IGF-1), IGF-1 gene expression, and IGF-1 binding proteins. J Gerontol 46:B180-B187 (1991).
152. Manjgaladze M, Chen S, Frame LT, Seng JE, Duffy PH, Feuers RJ, Hart RW, Leakey JEA. Effects of caloric restriction on rodent drug and carcinogen metabolizing enzymes: implications for mutagenesis and cancer. Mutat Res 295:201-222 (1993).
153. Prasanna HR, Lu MH, Hart RW. Status of dehydroepiandrosterone and hepatic metabolism of aflatoxin B1 in food restricted rats. Biochem Arch 6:419-427 (1990).
154. Bronson FH. Food-restricted, prepubertal, female rats: rapid recovery of luteinizing hormone pulsing with excess food, and full recovery of pubertal development with gonadotropin-releasing hormone. Endocrinology 118:2483-2487 (1986).
155. Holehan AM, Merry BJ. The control of puberty in the dietary restricted female rat. Mech Ageing Dev 32:179-191 (1985).
156. Bronson FH, Heideman PD. Short-term hormonal responses to food intake in peripubertal female rats. Am J Physiol 259:R25-R31 (1990).
157. Bronson FH, Marsteller FA. Effect of short-term food deprivation on reproduction in female mice. Biol Reprod 33:660-667 (1985).
158. Holehan AM, Merry BJ. Lifetime breeding studies in fully fed and dietary restricted female CFY Sprague-Dawley rats. 1: Effect of age, housing conditions and diet on fecundity. Mech Ageing Dev 33:19-28 (1985).
159. Chapin RE, Gulati DK, Fail PA, Hope E, Russell SR, Heindel JJ, George JD, Grizzle TB, Teague JL. The effects of feed restriction on reproductive function in Swiss CD-1 mice. Fundam Appl Toxicol 20:15-22 (1993).
160. Woodside B. Effects of food restriction on the length of lactational diestrus in rats. Horm Behav 25:70-83 (1991).
161. Young CM, Rasmussen KM. Effects of varying degrees of chronic dietary restriction in rat dams on reproductive and lactational performance and body composition in dams and their pups. Am J Clin Nutr 41:979-987 (1985).
162. Sisk CL, Bronson FH. Effects of food restriction and restoration on gonadotropin and growth hormone secretion in immature male rats. Biol Reprod 35:554-561 (1986).
163. Seng JE, Gandy J, Turturro A, Lipman R, Bronson RT, Parkinson A, Johnson W, Hart RW, Leakey JEA. Effects of caloric restriction on expression of testicular cytochrome P450 enzymes associated with the metabolic activation of carcinogens. Arch Biochem Biophys 335:42-52 (1996).
164. Johnson L, May MR, Busbee DL, Williams JD. Effect of age and dietary restriction on daily sperm production and number and transit time of epididymal spermatozoa in the mouse. Age 15:65-72 (1992).
165. Chapin RE, Gulati DK, Barnes LH, Teague JL. The effects of feed restriction on reproductive function in Sprague-Dawley rats. Fundam Appl Toxicol 20:23-29 (1993).
166. Thurman JD, Bucci T, Hart RW, Turturro A. Survival, body weight and spontaneous neoplasms in ad libitum fed and dietary restricted Fischer 344 rats. Toxicol Pathol 22:1-9 (1994).
167. Bronson FH. Relative effects of exercise, diet, and female stimulation on sexual aging of male mice. J Gerontol 37:555-559 (1982).
168. Klebanov S, Diais S, Stavinoha WB, Suh Y, Nelson JF. Hyperadrenocortism, attenuated inflammation, and the life-prolonging action of food restriction in mice. J Gerontol 50A:B78-B82 (1995).
169. Engelman RW, Day NK, Good RA. Calories, cell proliferation, and proviral expression in autoimmunity and cancer. Proc Soc Exp Biol Med 203:13-17 (1993).
170. Pashko LL, Schwartz AG. Reversal of food restriction-induced inhibition of mouse skin tumor promotion by adrenalectomy. Carcinogenesis 13:1925-1928 (1992).
171. Schwartz AG, Pashko LL. Role of adrenocortical steroids in mediating cancer-preventive and age-retarding effects of food restriction in laboratory rodents. J Gerontol 49:B37-B41 (1994).
172. Himeno Y, Engelman RW, Good RA. Influence of calorie restriction on oncogene expression and DNA synthesis during liver regeneration. Proc Natl Acad Sci USA 89:5497-5501 (1992).
173. Grasl-Kraupp B, Bursch W, Ruttkay-Nedecky B, Wagner A, Lauer B, Schulte-Hermann R. Food restriction eliminates preneoplastic cells through apoptosis and antagonizes carcinogenesis in rat liver. Proc Natl Acad Sci USA 91:9995-9999 (1994).
174. Muskhelishvili L, Hart RW, Turturro A, James SJ. Age-related changes in the intrinsic rate of apoptosis in livers of diet-restricted and ad libitum-fed B6C3F1 mice. Am J Pathol 147:20-24 (1995).
175. O'Steen WK, Landfield PW. Dietary restriction does not alter retinal aging in the Fischer 344 rat. Neurobiol Aging 12:455-462 (1991).
176. Green MW, Rogers PJ, Elliman NA, Gatenby SJ. Impairment of cognitive performance associated with dieting and high levels of dietary restraint. Physiol Behav 55:447-452 (1994).
177. Turturro A, Duffy PH, Hart RW. Modulation of toxicity by diet and dietary macronutrient restriction. Mutat Res 295:151-164 (1993).
178. Turturro A, Duffy P, Hart RW, Allaben WT. Rational use of dietary control in toxicity studies--B6C3F1 mouse. Toxicol Pathol 24:769-775 (1996).
179. Turturro A, Blank K, Murasko D, Hart R. Mechanisms of caloric restriction affecting aging and disease. Ann NY Acad Sci 719:159-170 (1994).
180. Bronson FH. Energy allocation and reproductive development in wild and domestic house mice. Biol Reprod 31:83-88 (1984).
181. Bronson FH. Mammalian reproductive strategies: genes, photoperiod and latitude. Reprod Nutr Dev 28:335-347 (1988).
182. Merry RJ, Holehan AM. The effect of dietary restriction on the endocrine control of reproduction. In: Biological Effects of Dietary Restriction (Fishbein L, ed). New York:Springer-Verlag, 1991;140-146.
183. Graves JL. The costs of reproduction and dietary restriction: parallels between insects and mammals. Growth Dev Aging 57:233-249 (1993).
184. Roe FJC. What does carcinogenicity mean and how should we test for it? Food Chem Toxicol 31:225-229 (1993).
185. Nohynek GJ, Longeart L, Geffray B, Provost JP, Lodole A. Fat, frail and dying young: survival, body weight and pathology of the Charles River Sprague-Dawley-derived rat prior to and since the introduction of the VAFR variant in 1988. Hum Exp Toxicol 12:87-98 (1993).
186. Keenan KP, Ballam GC, Dixit R, Soper KA, Laroque P, Mattson BA, Adams SP, Coleman JB. The effects of diet, overfeeding and moderate dietary restriction on Sprague-Dawley rat survival, disease and toxicology. J Nutr 127:851S-856S (1997).
187. Keenan KP, Smith PF, Hertzog P, Soper K, Ballam GC, Clark RL. The effects of overfeeding and dietary restriction on Sprague-Dawley rat survival and early pathology biomarkers of aging. Toxicol Pathol 22:300-315 (1994).
188. Edwards MJ, Keller BJ, Kauffman FC, Thurman RG. The involvement of Kupffer cells in carbon tetrachloride toxicity. Toxicol Appl Pharmacol 119:275-279 (1993).
189. Liu P, McGuire GM, Fisher MA, Farhood A, Smith CW, Jaeschke H. Activation of Kupffer cells and neutrophils for reactive oxygen formation is responsible for endotoxin-enhanced liver injury after hepatic ischemia. Shock 3:56-62 (1995).
190. Ishiyama H, Ogino K, Hobara T. Role of Kupffer cells in rat liver injury induced by diethyldithiocarbamate. Eur J Pharmacol 292:135-141 (1995).
191. Badger DA, Sauer J-M, Hoglen NC, Jolley CS, Sipes IG. The role of inflammatory cells and cytochrome P450 in the potentiation of CCl4-induced liver injury by a single dose of retinol. Toxicol Appl Pharmacol 141:507-519 (1996).
192. Allaben WT, Turturro A, Leakey JEA, Seng JE, Hart RW. Points-to-Consider Documents: the need for dietary control for the reduction of experimental variability within animal assays and the use of dietary restriction to achieve dietary control. Toxicol Pathol 24:776-781 (1996).
193. Beer SF, Bircham PMM, Bloom SR, Clark PM, Hales CN, Hughes CM, Jones CT, Marsh DR, Raggatt PR, Findlay ALR. The effect of a 72-h fast on plasma levels of pituitary, adrenal, thyroid, pancreatic and gastrointestinal hormones in healthy men and women. J Endocrinol 120:337-350 (1989).
194. Boyle PJ, Shah SD, Cryer PE. Insulin, glucagon, and catecholamines in prevention of hypoglycemia during fasting. Am J Physiol 256:E651-E661 (1989).
195. Thissen JP, Ketelslegers JM, Underwood LE. Nutritional regulation of the insulin-like growth factors. Endocrinol Rev 15:80-101 (1994).
196. Booth A, Mazur AC, Dabbs JM. Endogenous testosterone and competition: the effect of fasting. Steroids 58:348-350 (1993).
197. Hart RW, Keenan K, Turturro A, Abdo KM, Leakey JEA, Lyn-Cook B. Caloric restriction and toxicity: symposium overview. Fundam Appl Toxicol 25:184-195 (1995).
198. Colditz GA, Willett WC, Hunter DJ, Stampfer MJ, Manson JE, Hennekens CH, Rosner BA. Family history, age, and risk of breast cancer. Prospective data from the Nurses' Health Study. JAMA 270:338-343 (1993).
199. Gaziano JM, Manson JE. Diet and heart disease. The role of fat, alcohol, and antioxidants. Cardiol Clin 14:69-83 (1996).
200. Day GL, Blot WJ, Austin DF, Bernstein L, Greenberg RS, Preston-Martin S, Schoenberg JB, Winn DM, McLaughlin JK, Fraumeni JF Jr. Racial differences in risk of oral and pharyngeal cancer: alcohol, tobacco, and other determinants. J Natl Cancer Inst 85:465-473 (1993).
201. McLaughlin JK, Hrubec Z, Blot WJ, Fraumeni JF Jr. Smoking and cancer mortality among U.S. veterans: a 26-year follow-up. Int J Cancer 60:190-193 (1995).
202. Hart RW, Leakey JEA, Allaben WT, Chou M, Duffy PH, Feuers RJ, Turturro A. Role of nutrition and diet in degenerative processes. Int J Toxicol Environ Health 1:26-32 (1993).
203. Manson JE, Willett WC, Stampfer MJ, Colditz GA, Hunter DJ, Hankinson SE, Hennekens CH, Speizer FE. Body weight and mortality among women. New Engl J Med 333:677-685 (1995).
204. Willett WC, Manson JE, Stampfer MJ, Colditz GA, Rosner B, Speizer FE, Hennekens CH. Weight, weight change, and coronary heart disease in women. Risk within the 'normal' weight range. JAMA 273:461-465 (1995).
205. Ballard-Barbash R, Swanson CA. Body weight: estimation of risk for breast and endometrial cancers. Am J Clin Nutr 63:437S-441S (1996).
206. Ziegler RG, Hoover RN, Nomura AM, West DW, Wu AH, Pike MC, Lake AJ, Horn-Ross PL, Kolonel LN, Siiteri PK et al. Relative weight, weight change, height, and breast cancer risk in Asian-American women. J Natl Cancer Inst 88:650-660 (1996).
207. Chow WH, McLaughlin JK, Mandel JS, Wacholder S, Niwa S, Fraumeni JF Jr. Obesity and risk of renal cell cancer. Cancer Epidemiol Biomarkers Prev 5:17-21 (1996).
208. Shike M. Body weight and colon cancer. Am J Clin Nutr 63:442S-444S (1996).
209. Nauts HC. Bacterial pyrogens: beneficial effects on cancer patients. Prog Clin Biol Res 107:687-696 (1982).
210. McCord JM. Superoxide radical: controversies, contradictions and paradoxes. Proc Soc Exp Biol Med 209:112-117 (1995).
211. Vartanyan LS, Sadovnikova IP, Gurevich SM, Sokolova IS. Generation of superoxide radicals in membranes of subcellular organelles of regenerating liver. Biochem Engl Transl Biokhimiya 57:454-459 (1992).
212. Ziegler RG, Colavito EA, Hartge P, McAdams MJ, Schoenberg JB, Mason TJ, Fraumeni JF Jr. Importance of *-carotene, ß-carotene, and other phytochemicals in the etiology of lung cancer. J Natl Cancer Inst 88:612-615 (1996).
213. Kumari BS, Chandra RK. Overnutrition and immune responses. Nutr Res 13:S3-S18 (1993).
214. Prasad AK, Singh KP, Saxena AK, Mathur NK, Ray PK. Increased macrophage activity in protein A treated tumor regressed animals. Immunopharmacol Immunotoxicol 9:541-561 (1987).
215. Singh KP, Shau H, Gupta RK, Kopald K, Ray PK. Protein A potentiates lymphokine-activated killer cell induction in normal and melanoma patient lymphocytes. Immunopharmacol Immunotoxicol 14:79-103 (1992).
216. Nauts HC. Bacteria and cancer--antagonisms and benefits. Cancer Surv 8:713-723 (1989).
217. Tang ZY, Zhou HY, Zhao G, Chai LM, Zhou M, Lu JZ, Liu KD, Havas HF, Nauts HC. Preliminary result of mixed bacterial vaccine as adjuvant treatment of hepatocellular carcinoma. Med Oncol Tumor Pharmacother 8:23-28 (1991).
Last Update: March 11, 1998