Toxicological
Defense Mechanisms and the Shape of Dose-Response Relationships
Environmental
Health Perspectives 106, Supplement 1, February 1998
Division of Toxicology, Agency for Toxic Substances and Disease Registry, Atlanta, Georgia
Key words: ATSDR, risk analysis, toxicokinetics, mechanisms, adaptation, toxicodynamics, hormesis, BMD, PBPK, mixtures
This paper is based on a presentation at The Third BELLE Conference on Toxicological Defense Mechanisms and the Shape of Dose-response Relationships held 12-14 November 1996 in Research Triangle Park, NC. Manuscript received at EHP 11 March 1997; accepted 2 June 1997.
The authors thank N. Haynie-Mooney and E. Julian for their assistance in preparation of this manuscript.
Address correspondence to Dr. D.E. Jones, Agency for Toxic Substances and Disease Registry, MS-E29, 1600 Clifton Road, Atlanta, GA 30333. Telephone: (404) 639-4605. Fax:(404) 639-6316. E-mail: dej2@cdc.gov
Abbreviations used: ADI, acceptable daily intake; ATSDR, Agency for Toxic Substances and Disease Registry; BMD, benchmark dose; CDD, chlorinated dibenzodioxin; CDF, chlorinated dibenzofuran; CNS, central nervous system; ESADDI, estimated safe and adequate daily intake; ETE, essential trace element(s); LOAEL, lowest observed adverse effect level; MRL, minimal risk level; MTBE, methyl-t-butyl ether; NOAEL, no observed adverse effect level; NPL, National Priorities List; PBPK, physiologically based pharmacokinetic; PCE, tetrachloroethylene; ppm, parts per million; RfCs, reference concentrations; RfDs, reference doses; TCA, trichloroacetic acid; TCDD, 2,3,7,8-tetrachlorodibenzo-p-dioxin; TCE, trichloroethylene; TEF, toxicity equivalency factor(s); UF, uncertainty factor; U.S. EPA, Environmental Protection Agency; U.S. FDA, U.S. Food and Drug Administration; WOE, weight of evidence.
The ATSDR, like other agencies such as the U.S. Environmental Protection Agency (U.S. EPA) and the U.S. Food and Drug Administration (U.S FDA), has relied on risk assessment in its task of assessing the public health implications of low-level exposures to hazardous substances. As defined by the National Academy of Sciences in 1983, risk assessment consists of four interrelated steps or components: hazard identification, dose-response assessment, exposure assessment, and risk characterization (2). In the real-life risk assessment process, the fourth step, risk characterization, is actually an integration of the other three components into a qualitative and/or quantitative assessment that characterizes the probability of adverse health effects in an exposed population. The ATSDR approach for risk characterization of noncancer adverse health effects is primarily a qualitative rather than a probabilistic undertaking. Nevertheless, the ATSDR does use minimal risk levels (MRLs), health guidance values developed by the ATSDR specifically to identify levels of exposure thought to be without appreciable risk over specified durations and routes of exposure (3). Although the procedures used to derive such health guidance values by agency scientists is operationally straightforward, the judgment that goes into development of these values is guided by a full range of expert judgment in the fields of toxicology, epidemiology, and pathology, as detailed below.
In this paper we discuss the role of health guidance values in public health practice and the application of mechanistic insights in deriving health guidance values. Three questions posed are vital to how regulatory and public health agencies consider the biological effects of low-level exposures (4):
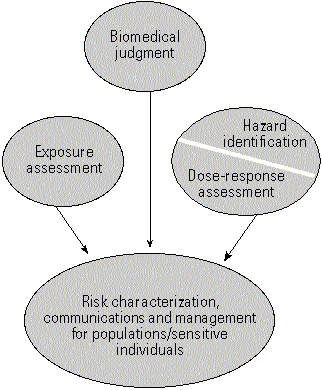
As applied by the ATSDR in public health practice, risk analysis is a multidimensional endeavor encompassing the concepts of risk assessment, biomedical judgment, risk management, and risk communication (Figure 1). Its application can be visualized within the broader context of the agency's public health assessment program (Figure 2). As part of its public health mission, the ATSDR develops public health assessments to evaluate the public health implications of all hazardous waste sites on the National Priorities List (NPL) and for selected sites identified by public petition (1). The ATSDR's public health assessments are based on three key components: environmental monitoring data, health outcome data, and community health concerns. These three components are then evaluated in the context of what is known about the toxicity of site-related contaminants, the probability of past, present, or future exposures, and the significance of site-specific conditions, including demographic and other parameters of exposure. In these evaluations the ATSDR may use health guidance values specifically to determine which chemicals should be addressed further in terms of risk posed to exposed communities based on a comparison with environmental monitoring data. Depending on the outcome of the ATSDR analysis, a number of follow-up activities might be considered. Possible follow-up activities may include further health study and/or surveillance, health education, research, and the development of chemical-specific exposure registries.
Figure 1. Risk analysis as a multidimensional endeavor, encompassing the components of risk assessment, biomedical judgement, risk management, and risk communication.

Figure 2. Application of the concept of risk analysis to the ATSDR public health assessment program.
These MRLs, their supporting databases, and an explanation of factors considered in their derivation are included in toxicological profiles developed by the ATSDR (7). Procedurally, the derivation of an MRL is straightforward and analogous to the derivation of acceptable daily intakes (ADIs) and reference doses/reference concentrations (RfDs/RfCs) developed by the U.S. FDA and the U.S. EPA, respectively. Specific internal guidance for derivation of MRLs by the ATSDR has been published (3).
Traditionally, health guidance values have been derived from either controlled human clinical studies, human epidemiologic studies (usually retrospective), or controlled studies involving laboratory animals serving as surrogates for human populations. Based on a review of these studies, the lowest dose or exposure level at which an effect considered adverse was observed (LOAEL) and the highest level below which no adverse effect was observed (NOAEL) is similarly determined. Because the population of interest in the health study typically is not the same as a potentially exposed population to be protected by the health guidance value, mathematical adjustments are made to the LOAEL or NOAEL to express uncertainties inherent in the assumptions and database used to calculate the health guidance value.
The uncertainty factors (UFs) proposed by Barnes and Dourson (8) and presented in Table 1 provide the basis for the approach and types of mathematical adjustments used by the ATSDR in deriving its MRLs. Each MRL thus has its own area of uncertainty with regard to its derivation based primarily on the study used as the basis for deriving the MRLs. The aggregate UFs can be viewed as loose upper-bound estimates that account for differences in susceptibility between test and target species and for sensitivity differences within the human population. The overall magnitude of the UF reflects the confidence in the final calculated number and the database supporting that number for a given chemical. As a result the final health guidance value, or MRL, can be viewed as an estimate of a dose that is likely to be without adverse effects in sensitive individuals for a specified duration and route of exposure.

Mathematically the derivation of the MRL can be expressed as follows:
![]()
where MF=a modifying factor and the NOAEL, the LOAEL, and the UF are as previously defined.
This formula provides a computational method for determining a reference value for a particular substance and a particular route and duration of exposure. MRLs thus derived provide route-specific guidance that would protect all potentially exposed populations. As such, they also provide a basis for determining screening or trigger levels for chemical-specific exposures of concern on a site-specific basis. Health guidance values such as MRLs, RfDs/RfCs, and ADIs can all be used in specific exposure scenarios to estimate levels of a substance in a particular environmental medium that can serve as a basis to determine whether further evaluation is warranted. Such health guidance values as used by the ATSDR are intended to be used only as screening tools. They are not intended to be interpreted as precise values or used as action levels but as indicators of whether further evaluation is warranted for a particular exposure scenario. Moreover, because of the specific exposure route, exposure duration, and toxicity end point associated with each health guidance value, the values can also be used to alert health care providers about what outcomes they should be concerned with in a particular locality or site vicinity.
Toxicokinetic Mechanisms. Several examples in which the ATSDR used a knowledge of chemical-specific toxicokinetic mechanisms in derivation of its MRLs are listed and discussed below. These examples are summarized in Table 2.
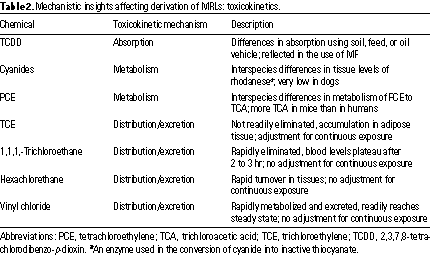
Absorption/Bioavailability. 2,3,7,8-TETRACHLORODIBENZO-P-DIOXIN (TCDD). The ATSDR has developed a proposed acute oral MRL for TCDD based on a study in mice exposed to 14 daily doses of TCDD by gavage in oil vehicle (9). An important issue considered during derivation of this MRL was the bioavailability of TCDD (10): It has been well documented in animal studies that adsorption of TCDD from the gastrointestinal tract depends largely on the carrier vehicle (11). Absorption of TCDD from both soil and feed has been estimated to be 20 to 60%, whereas the adsorption of TCDD from a corn oil vehicle has been estimated to be as much as 85%. Because the likely means of environmental exposure to TCDD is through ingestion of contaminated food or soil but the definitive animal TCDD toxicity study used corn oil gavage, MRL derivation was divided by an additional modifying factor of 0.5 to adjust for the approximate 2-fold greater bioavailability from corn oil compared to that from food or soil.
Metabolism. Many toxic chemicals are metabolized by similar metabolic pathways across species. However, interspecies differences are also well documented (12). These differences often are attributed to lack of particular enzymes or differences in the relative importance of particular routes of metabolism. Cyanides and tetrachloroethylene (PCE) are examples of chemicals with specific mechanistic differences in metabolism that affected derivation of the ATSDR MRL.
CYANIDES. Rhodanese is an enzyme used in the conversion of cyanide into inactive thiocyanate. Dog tissues have low levels of rhodanese compared with levels in other species; therefore dogs are extremely susceptible to cyanide poisoning. As a result, cyanide metabolism in a species such as the rat more accurately reflects cyanide metabolism in the human than does cyanide metabolism in the dog. Because of this, the ATSDR based its intermediate-duration oral MRL on a study in rats even though a similar study in dogs reported much lower toxic effect levels (13).
TETRACHLOROETHYLENE. During the ATSDR derivation of MRLs for intermediate-duration inhalation and oral exposures to PCE, it was noted that the proposed toxic end point reported for mice (peroxisomal proliferative response in the liver) might not be relevant to humans. Several differences in PCE metabolism exist between mice and humans. The rate of metabolism of PCE to trichloroacetic acid (TCA)--the reactive metabolite in the hepatic toxicity--is much greater in mice than in humans (14). Moreover, mice and rats also respond to TCA and many other chemicals by induction of hepatocellular peroxisomes, whereas humans either are much less responsive to peroxisome proliferators or do not respond at all to doses that cause marked responses in rodents. Because humans produce little TCA following PCE exposure and because peroxisomal proliferation in humans is minimal compared to that in rodents, liver hypertrophy and tumor development as observed in mice may not occur by the same mechanism in humans or may be much less likely to occur. Therefore, the ATSDR did not derive intermediate-duration MRLs for PCE from these rodent studies but instead deferred derivation until the significance of these mechanistic differences between humans and rodents is more fully understood.
Distribution and Excretion. According to the ATSDR MRL methodology (3), if a study serving as the base for an MRL derivation does not involve continuous dosing over the entire exposure period, an adjustment can be made to correct for intermittent exposure. Generally, the intermittent exposure dose is multiplied by correction factors to adjust for a full 24 hr/day and 7 days/week of exposure.
More specifically, if study results indicate that a toxic end point depends directly on the duration of exposure (e.g., if metabolism and excretion is moderate to slow or the study results indicate that the toxic end point is a cumulative effect), adjustments to correct for intermittent exposure may be appropriate. Conversely, if the critical effects are considered to depend mainly on exposure concentration rather than length of exposure and if the substance being tested is rapidly metabolized and/or eliminated, dose adjustment for intermittent exposure generally is considered inappropriate. A number of examples are presented below in which the ATSDR considered the appropriateness of dose adjustment for intermittent exposure in derivation of MRLs.
TRICHLOROETHYLENE. An acute-duration inhalation MRL was derived for trichloroethylene (TCE) based on neurologic effects observed in human volunteers (15). An intermediate-duration inhalation MRL was also derived from a study in which neurological effects were seen in rats (16). Because the toxicokinetic data indicate that TCE is not readily eliminated and can accumulate in adipose tissue (17), both MRLs were adjusted for intermittent exposure.
1,1,1-TRICHLOROETHANE. An acute-duration inhalation MRL was derived for 1,1,1-trichloroethane based on an LOAEL for reduced performance on psychomotor tests reported in humans (18). However, 1,1,1-trichloroethane is rapidly eliminated in the expired air and blood levels of 1,1,1-trichloroethane plateau after 2 to 3 hr (18). These findings suggest that longer exposure would not lead to higher body burdens. Therefore, a correction for intermittent exposure was considered inappropriate for the acute-duration inhalation MRL for 1,1,1-trichloroethane (19).
HEXACHLOROETHANE. An acute-duration inhalation MRL was derived for hexachloroethane based on an NOAEL for neurological effects reported in pregnant rats exposed to hexachloroethane (20). Laboratory data showed that effects depend on blood levels, which directly depend on concentration, not duration of treatment (21). Moreover, the toxicokinetic data also showed a rapid turnover in the tissues. Therefore, no adjustment was made to the MRL to correct for intermittent exposure.
VINYL CHLORIDE. An acute-duration inhalation MRL was derived for vinyl chloride based on developmental effects in offspring of mice exposed during gestation (22,23). An intermediate-duration inhalation MRL was also derived based on hepatic effects in rats (24). A review of the toxicokinetic data indicated that vinyl chloride readily reaches steady state, is rapidly metabolized and excreted, and neither it nor its metabolites readily accumulate in tissue (25). Therefore, no adjustment for intermittent exposure was made in the derivation of either the acute or the intermediate-duration inhalation MRL for vinyl chloride.
Toxicodynamic Mechanisms. Chemical interactions with specific receptors or enzymes are examples of toxicodynamic mechanisms. Such mechanisms and their associated interspecies differences may play critical roles in the expression of toxicity as well as in deliberations about risk assessment for toxic chemicals. Two specific examples of how understanding such toxicodynamic mechanisms has affected the ATSDR derivation of MRLs are discussed below.
Chlorinated dibenzodioxins and dibenzofurans. Chlorinated dibenzodioxins (CDDs) and chlorinated dibenzofurans (CDFs) are groups of halogenated aromatic hydrocarbons that are closely related chemically and often occur together in the environment (26). To estimate the risk associated with exposure to chemical congeners of these two closely related chemical classes, the ATSDR has adopted the U.S. EPA toxicity equivalency factors (TEF) method, which is based on congener-specific data and the assumption that Ah receptor-mediated toxicity is common to dioxinlike congeners and is additive (27). The TEF scheme compares the relative toxicity of individual CDD and CDF congeners to that of 2,3,7,8-TCDD, the most toxic and extensively studied of these halogenated aromatic hydrocarbons. The TEF for 2,3,7,8-TCDD is defined as unity, whereas TEF values for all other CDD and CDF congeners are less than 1 (0 has been assigned to all non-2,3,7,8-substituted congeners); this reflects the lower toxic potency of most CDD and CDF congeners. A description of the ATSDR MRL values for a number of CDDs and CDFs using the TEF approach was recently published in an article that also points out a number of uncertainties associated with this method (28).
METHYL-T-BUTYL ETHER. An MRL for chronic-duration
inhalation exposure to methyl-t-butyl ether (MTBE) was derived from
an NOAEL for chronic progressive nephropathy in female rats (29). Chronic
progressive nephropathy is an age-related spontaneous disorder of rats
that is more severe in males than in females and affects certain strains
more than others (30). Chronic exposure of male rats to ![]() 2µ-globulin-inducing
agents results in aggravation of chronic progressive nephropathy, which
is characterized by increased severity and earlier onset of the disease.
Based on these considerations, the ATSDR concluded that the higher incidence
and greater severity of chronic progressive nephropathy observed at lower
MTBE exposure concentrations in male rats compared with that in female
rats may have been due to exacerbation of this syndrome by the accumulation
of
2µ-globulin-inducing
agents results in aggravation of chronic progressive nephropathy, which
is characterized by increased severity and earlier onset of the disease.
Based on these considerations, the ATSDR concluded that the higher incidence
and greater severity of chronic progressive nephropathy observed at lower
MTBE exposure concentrations in male rats compared with that in female
rats may have been due to exacerbation of this syndrome by the accumulation
of ![]() 2µ-globulin
or another unknown protein unique to male rats. As a result this end point
was considered unsuitable for MRL derivation. However, because female rats
also exhibited enhanced chronic progressive nephropathy but at higher exposure
levels, the NOAEL for this end point in female rats was used (instead of
the NOAEL observed in male rats) to derive the chronic-duration inhalation
MRL for MTBE.
2µ-globulin
or another unknown protein unique to male rats. As a result this end point
was considered unsuitable for MRL derivation. However, because female rats
also exhibited enhanced chronic progressive nephropathy but at higher exposure
levels, the NOAEL for this end point in female rats was used (instead of
the NOAEL observed in male rats) to derive the chronic-duration inhalation
MRL for MTBE.
Adaptive responses within organisms may involve biochemical or structural alterations. Examples of adaptive biochemical responses include induction of the cytochrome P450 mixed-function oxidase system in the liver or other organs as well as glutathione depletion and synthesis in the liver. Examples of structural adaptive responses within tissues include atrophy, hypertrophy, hyperplasia, and metaplasia. Adaptive responses generally are considered to enhance an organism's performance or its ability to withstand challenge. Actually, however, some alterations that typically are classified as adaptive responses may have potentially harmful (i.e., adverse) effects on the host in some cases. An adverse effect is defined as "any effect that reduces the capacity of an organism or a component of the organism to function in a normal manner or diminishes the ability to withstand further stress" (31).
Classification of biologic changes in tissues often is challenging. For example, the borders between hyperplasia and neoplasia and between benign and malignant neoplasia often are indistinct. Similarly, the boundary between adaptive and toxic responses often is not well delineated. As a result biologic assessment of adaptive responses as adverse or not adverse is a matter of judgment and sometimes is controversial. Hypertrophy of skeletal muscle in response to increased workload is an example of an adaptive change that might prove beneficial to the host. However, hypertrophy of the left heart ventricle because of arterial hypertension, even though it allows the heart to function against an increased workload, will result in decreased cardiac ability to compensate for additional stress. Metaplasia also typically is considered an adaptive response but the predictive value for lesion progression and secondary effects is not always clear. If metaplasia occurs in the pancreas (e.g., squamous metaplasia of pancreatic ducts associated with exposure to a test substance), this process generally does not interfere with pancreatic function. However, if squamous metaplasia occurs in the tracheal epithelium it may interfere with normal respiratory defense function (mucociliary escalator).
A common biochemical adaptive response is induction of the cytochrome P450 mixed-function oxidase system (32-34). Many chemicals evaluated by the ATSDR (e.g., aldrin, chloroform, DDT, PCE) can induce the cytochrome P450 system in the liver or other organs. This induction leads to stimulation of protein synthesis and proliferation of smooth endoplasmic reticulum. Depending on the inducing agent, one or more isoenzymes of P450 may be induced, each of which has different affinities for a variety of other substances. The P450 system plays a critical role in the metabolism of both endogenous and exogenous compounds. In some cases, as in acetaminophen or carbon tetrachloride, this can be viewed as a toxification reaction (i.e., the parent compound is metabolized to a more toxic species). In other cases, as with ethanol or phenobarbital, the cytochrome P450 system detoxifies the parent compound, which results in less reactive metabolites. However, once induction of the mixed-function oxidase system has occurred (no matter what chemical was the initiater), this adaptive response has significant implications in future chemical exposures. This adaptive modification may potentiate or inhibit toxic responses to subsequent chemical exposures. The enhancement or inhibition of a compound's metabolism can lead to toxicological interactions that may be important in site-specific risk assessments (35). This concept is particularly significant to the ATSDR in its assessment of NPL sites, where multichemical exposures are much more likely than exposure to a single chemical.
Within the framework of human health risk assessment, hepatic cytochrome P450 induction generally should be classified as an adverse effect. This induction alters the normal functioning of the organ (i.e., the organ is able to metabolize compounds at a greater rate). Even though this effect may be beneficial for some compounds it may be detrimental for others. Additionally, the designation of P450 induction as an adverse effect reflects the judgment that involuntary environmental exposure to chemicals should not be at levels that alter the normal state of the organism.
Glutathione depletion in the liver is another example of a biochemical adaptive response. The hepatic metabolism of chemicals such as acetaminophen can result in such depletion. Acetaminophen is metabolized by cytochrome P450 to a reactive intermediate that is detoxified by conjugation with glutathione (36). As long as levels of glutathione are not depleted, no hepatotoxic effects are induced. However, high doses of acetaminophen result in glutathione depletion, after which overt toxic effects are likely in the liver. Such chemical-induced depletion of liver glutathione is considered an adverse effect because it diminishes the normal capacity of the liver to respond to other chemical agents.
Clearly, P450 induction and glutathione depletion are not frank toxic effects. At the whole-organism or whole-organ levels no damage may be apparent. Consequently, adaptive or compensatory changes such as P450 induction or hepatic glutathione depletion are most appropriately classified as minimal adverse effects. If exposures causing such effects are associated with more severe responses, the nature of these responses rather than the associated adaptive changes should be used to evaluate toxicity at that level of exposure.
To be consistent with the ATSDR's public health mission of preventing and mitigating adverse health effects caused by exposure to hazardous substances, it is important to assess adaptive responses to chemical exposures on a case-by-case basis. Within the definitions presented earlier and the general context of risk analyses involving chemical contamination, an adaptive response may be classified as adverse when the adaptive alteration potentially contributes to frank toxic effects, when it diminishes the organism's ability to withstand further stress, or when it compromises an organism's normal ability to dispose of chemical substances.
All these criteria are individually and collectively considered by the ATSDR in assessing the risk posed by exposure to toxic substances. Adaptive response may actually be the basis for selecting the toxic end points the ATSDR uses in its derivation of health guidance values. Moreover, the understanding and interpretation of such adaptive responses may also affect how the agency evaluates multiple chemical exposures at complex waste sites. Clearly, an in-depth understanding of adaptive mechanisms is crucial to the ATSDR's multifaceted risk-assessment process.
As previously mentioned, the ATSDR carefully considers the biological effects of low-level exposures in its human health assessments, particularly in those deliberations involving the derivation of MRLs. Any evidence of hormesis (i.e., the induction of beneficial effects by low doses of otherwise harmful physical or chemical agents) has always been carefully noted and considered in these deliberations. However, even though the concept of hormesis has been the subject of extensive scientific debate in recent years (40,41), it still is not known whether hormetic effects occur with most toxicological end points (e.g., carcinogenesis, immunotoxicity, mutagenesis, teratogenesis) or whether hormetic effects occur at the same site of action for both low and high levels of exposure.
Nonetheless, the fact remains that many chemicals on the ATSDR list of hazardous substances known to be toxic at high exposure levels can cause deficiency associated toxicity if exposure levels are extremely low or absent (e.g., chromium, manganese, zinc). For each substance in its MRL deliberations the ATSDR considers not only high-dose toxicity but also any evidence of essentiality or beneficial effects as well as any evidence of deficiency associated toxicity.
The best known and understood chemical substances identified as beneficial at extremely low doses are dietary essential trace element(s) (ETE), i.e., chemical substances that must be present in small quantities in the human diet to maintain normal physiological functions. For each ETE, two ranges of exposure or intake are associated with adverse health effects: intakes that are too low and result in nutritional deficiency and intakes that are too high and cause toxicity. Some examples of the types of information considered by the ATSDR in derivation of oral MRLs for a number of ETEs are discussed below.
In a number of animal studies severe Cr deficiency in animal studies has resulted in hyperglycemia, decreased weight gain, elevated serum cholesterol levels, aortic plaques, corneal opacities, impaired fertility, and lethality (43). For humans the estimated safe and adequate daily intake (ESADDI) is 50 to 200 µg (44). In high-dose studies in mice with either chromium sulfate or potassium dichromate, the most sensitive toxic end point observed was decreased spermatogenesis (45). However, this end point was classified by the ATSDR as a serious effect and therefore inappropriate for derivation of an MRL (3,43). Because of the lack of data appropriate for deriving an MRL, the ATSDR adopted the upper limit of the ESADDI--200 µg Cr/day--as an interim health guidance value for Cr(III) and Cr(aVI). Such guidance was deemed both appropriate and necessary because of the prevalence of Cr at hazardous waste sites, the fairly complete database on Cr, and the fact that Cr is an essential nutrient.
The ESADDI for Mn intake for adults is 2 to 5 mg/day (44). Because appropriate data are lacking, no oral MRLs have been derived for either acute-, intermediate-, or chronic-duration exposure to Mn. As a result the ESADDI upper limit of 5 mg/kg/day was used to derive an interim health guidance value of 0.07 mg/kg/day until such time that information appropriate for derivation of these MRLs becomes available (49). The ATSDR considers such interim guidance both appropriate and necessary because of the prevalence of Mn at hazardous waste sites and the fact that Mn is an essential nutrient.
The ATSDR derived an intermediate oral MRL of 0.3 mg Zn/kg/day based on hematologic effects, including decreased hematocrit, serum ferritin, and erythrocyte superoxide dismutase activity, in women given daily supplements of Zn as Zn gluconate for 10 weeks (54,61). Normally for derivations of this type a UF of 100 would have been used (10 for sensitive human populations and 10 for the use of an LOAEL instead of an NOAEL) (3). However, because this LOAEL was considered to be a minimal LOAEL (3,61) and because Zn is an essential nutrient, a total UF of 3 was used instead of the normally applied 100. The 0.3 mg Zn/kg/day MRL thus derived has also been adopted as a chronic oral MRL because of a lack of adequate long-term studies in either humans or animals. This value is expected to be without adverse effects when Zn is consumed at this level on a daily basis over a long period of time and neither induces nutritional deficiency in healthy, nonpregnant adults ingesting the average American diet nor causes toxicity.
If toxicity is attributable to a stable metabolite, however, the current ATSDR MRL methodology may actually underestimate the relative human hazard. For example, the ATSDR's current acute oral MRL of 0.5 mg/kg/day for TCE was derived from an LOAEL for developmental effects in mice (66) divided by a UF of 100 (10 for use of an LOAEL and 10 for animal to human extrapolation) (17). In comparison, PBPK modeling of TCE (67) provided dose metrics for the area under the concentration curve for both the parent chemical and the metabolite, TCA. Dividing these dose metrics by a UF of 30--10 for use of an LOAEL and 3 for animal-to-human extrapolation (instead of 10 because of the reduced uncertainty provided by pharmacokinetic modeling)--an acute oral MRL for TCE was calculated to be either 0.02 mg/kg/day based on the TCE dose metric or 0.04 mg/kg/day based on the TCA dose metric. Because the TCA metabolite tends to accumulate in the fetus through an ion-trapping mechanism and acts as a teratogen by coagulating fetal proteins, this would support the application of PBPK modeling to lower the acute oral MRL for TCE.
The ATSDR's current intermediate oral MRL of 0.002 mg/kg/day for inorganic mercury was based on an NOAEL for increased kidney weight in rats divided by a UF of 100 (10 for animal-to-human extrapolation and 10 for human variability) (71). In a BMD analysis of inorganic mercury studies recently conducted by the ATSDR, relative kidney weight was used as the response to calculate BMDs from the same study using the Weibull model. Application of 100-fold UF for intra- and interspecies variability yielded the following BMD-based intermediate oral MRL for inorganic mercury: 0.003 mg/kg/day for an estimated 10% risk, 0.002 mg/kg/day for an estimated 5% risk, and 0.0003 mg/kg/day for an estimated 1% risk.
In this case, the BMD-based intermediate oral MRL for an estimated 5% risk was the same as the ATSDR's current MRL.
Qualitative (type of toxicity) and semiquantitative (direction of response not specific magnitude) evaluations have been prepared on binary weight-of-evidence combinations of chemicals included in two four-component mixture studies conducted by the TNO. These evaluations were used to estimate the overall toxicities of the mixtures that were then compared with experimentally determined toxicities (74,75). Preliminary analyses of the data indicate that the WOE approach quantitatively accounts for the observed interactions for mixtures of similarly acting renal toxicants, but for dissimilarly acting renal toxicants the method performs less well. This could be attributed to the fact that WOE evaluations are based on dose additivity that postulates that all chemicals in a given mixture act in the same way by the same mechanism and differ only in their potencies. Therefore, although this approach may be inappropriate for evaluating interactions for dissimilar acting agents, it may hold promise as a means to identify agents having different mechanisms of action.
References
1. ATSDR. Statement of organization, functions, and delegations of authority. Fed Reg 54(156):33617 (1989).
2. National Academy of Sciences. Risk Assessment in the Federal Government: Managing the Process. Washington:National Academy Press, 1983.
3. ATSDR. Minimal risk levels for priority substances and guidance for derivation; republication. Fed Reg 61(125): 33511-33520 (1996).
4. Biological Effects of Low-Level Exposures. Announcing the Third BELLE Conference on Toxicological Defense Mechanisms and the Shape of Dose-Response Relationships. BELLE Newslett 5(1):17-19 (1996).
5. CEQ. Risk Analysis: a guide to principles and methods for analyzing health and environmental risks. NTIS PB89-137772. Washington:Council on Environmental Quality, 1989.
6. De Rosa CT, Stevens Y-W, Johnson BJ. Cancer policy framework for: health assessment of carcinogens in the environment. Toxicol Ind Health 9(4):559-575 (1993).
7. De Rosa CT. Agency for Toxic Substances and Disease Registry's toxicological profiles: contribution to public health. Toxicol Ind Health 10(3):117 (1994).
8. Barnes DG, Dourson M. Reference dose (RfD): description and use in health risk assessment. Regul Toxicol Pharmacol 8:471-486 (1988).
9. White KL JR, Lysy HH, McCay JA, Anderson JC. Modulation of serum complement levels following exposure to polychlorinated-p-dioxins. Toxicol Appl Pharmacol 84:209-219 (1987).
10. Pohl H, De Rosa C, Holler J. Public health assessment for dioxin exposure from soil. Chemosphere 31(1):2437-2454 (1995).
11. Poiger H, Schlatter CH. Influence of solvents and absorbants on dermal and intestinal absorption of TCDD. Food Cosmet Toxicol 18:471-481 (1980).
12. Office of Technology Assessment. Researching Health Risks. OTA-BBS-570. Washington:U.S. Government Printing Office, 1993.
13. ATSDR. Toxicological Profile for Cyanide. Atlanta, GA:Agency for Toxic Substances and Disease Registry, 1996.
14. ATSDR. Toxicological Profile for Tetrachloroethylene. Atlanta, GA:Agency for Toxic Substances and Disease Registry, 1995.
15. Stewart RD, Dodd HC, Gay HH, Erley DS. Experimental human exposure to trichloroethylene. Arch Environ Health 20:64-71 (1970).
16. Arito H, Takahashi M, Ishikawa T. Effect of subchronic inhalation exposure to low-level trichloroethylene on heart rate and wakefulness-sleep in freely moving rats. Sangyo Igaku 36:1-8 (1994).
17. ATSDR. Toxicological Profile for Trichloroethylene. Atlanta, GA:Agency for Toxic Substances and Disease Registry, 1995.
18. Mackey CJ, Campbell L, Samuel AM, Alderman KJ, Idzikowski C, Wilson AK, Gompertz D. Behavioral changes during exposure to 1,1,1-trichloroethane: time-course and relationship to blood solvent levels. Am J Ind Med 11:223-240 (1987).
19. ATSDR. Toxicological Profile for 1,1,1,-Trichloroethane. Atlanta, GA:Agency for Toxic Substances and Disease Registry, 1995.
20. Weeks MH, Angerhofer RA, Bishop R, Thomasino J, Pope CR. The toxicity of hexachloroethane in laboratory animals. Am Ind Hyg Assoc J 40:187-199 (1979).
21. ATSDR. Toxicological Profile for Hexachloroethane. Atlanta, GA:Agency for Toxic Substances and Disease Registry, 1994.
22. John JA, Smith FA, Leong BKJ, Schwetz BA. The effects of maternally inhaled vinyl chloride on embryonal and fetal development in mice, rats, and rabbits. Toxicol Appl Pharmacol 39(3):497-513 (1977).
23. John JA, Smith FA, Schwetz BA. Vinyl chloride: inhalation teratology study in mice, rats, and rabbits. Environ Health Perspect 41:171-177 (1981).
24. Bi WF, Wang YS, Huang MY, Meng DS. Effects of vinyl chloride on testis in rats. Ecotoxicol Environ Safety 10:281-289 (1985).
25. ATSDR. Toxicological Profile for Vinyl Chloride. Atlanta, GA:Agency for Toxic Substances and Disease Registry, 1995.
26. ATSDR. Toxicological Profile for Chlorodibenzofurans. Atlanta, GA:Agency for Toxic Substances and Disease Registry, 1994.
27. U.S. EPA Risk Assessment Forum. Interim Procedures for Estimating Risks Associated with Exposure to Mixtures of Chlorinated Dibenzo-p-dioxins and Dibenzofurans (CDDs and CDFs) and 1989 Update. EPA 625/3-89/016. Washington:U.S. Environmental Protection Agency, 1989.
28. Pohl H, Holler J. Halogenated aromatic hydrocarbons and toxicity equivalency factors (TEFs) in the public health assessment perspective. Chemosphere 31(1):2547-2559 (1995).
29. Chun JS, Burleigh-Flayer HD, Kintigh WJ. Methyl Tertiary Butyl Ether: Vapor Inhalation Oncogenicity Study in Fischer 344 Rats. Project No 91N0013B. Export, PA:Bushy Run Research Center, 1992.
30. Williams MD, Durkin P. Unpublished data.
31. ATSDR. Toxicological Profile for Methyl-t-Butyl Ether (MTBE). Atlanta, GA:Agency for Toxic Substances and Disease Registry, 1994.
32. Loomis TA. Essentials of Toxicology. 3rd ed. Philadelphia:Lea and Febiger Press, 1978;245.
33. Kyle ME, Farber JL. Biochemical mechanisms of toxic cell injury. In: Handbook of Toxicologic Pathology (Haschek WM, Rosseaux CG, eds). New York:Academic Press, 1991;71-90.
34. Plaa GL. Toxic responses of the liver. In: Casarett and Doull's Toxicology. 3rd ed. (Klaassen CD, Amdur MO, Doull J, eds). New York:MacMillan, 1986;334-353.
35. Mumtaz MM, De Rosa C, Durkin PR. Approaches and challenges in risk assessments of chemical mixtures. In: Toxicology of Chemical Mixtures (Yang RSH, ed). San Diego:Academic Press, 1994;565-597.
36. Connolly RB. U.S. EPA reassessment of the health risks of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). CIIT Activities 14(12):1-8 (1994).
37. Agency for Toxic Substances and Disease Registry. Revised priority list of hazardous substances that will be the subject of toxicological profiles. Fed Reg 61:18744-18745 (1996).
38. Calabrese EJ, McCarthy ME, Kenyon E. The occurrence of chemically induced hormesis. Health Phys 52(5):531-541 (1987).
39. Calabrese EJ, Baldwin L. Possible examples of chemical hormesis in a previously published study. J Appl Toxicol 13(3):169-172 (1993).
40. Sagan L, ed. The Conference on Radiation Hormesis. Oakland, CA, 14-16 August 1985. Proceedings. Health Phys 52:519-678 (1987).
41. Liu S-Z, ed. Proceedings of the International Symposium on Biological Effects of Low Level Exposures to Radiation and Related Agents, Changchun, China. Princeton, NJ:Princeton Scientific Publishers, 1994.
42. International Programme on Chemical Safety. Chromium. Environmental Health Criteria 61. Geneva:World Health Organization, 1988.
43. ATSDR. Toxicological Profile for Chromium. Atlanta, GA:Agency for Toxic Substances and Disease Registry, 1993.
44. National Research Council. Recommended Dietary Allowances. 10th ed. Washington:National Academy Press, 1989.
45. Zahid ZR, Al-Hakkak ZS, Kadhim AHH, Elias EA, Al-Jumaily IS. Comparative effects of trivalent and hexavalent chromium on spermatogenesis of the mouse. Toxicol Environ Chem 25:131-136 (1990).
46. Wedler FC. Biochemical and nutritional role of manganese: an overview. In: Manganese in Health and Disease (Klimis-Tavantzis DJ, ed). Boca Raton, FL:CRC Press, 1994;1-36.
47. Keen CL, Lonnerdal B, Hurley LS. Manganese. In: Biochemistry of the Essential Ultratrace Elements (Frieden E, ed). New York:Plenum Press, 1984;89-132.
48. WHO. Trace elements in human nutrition; Report of a WHO Expert Committee. WHO Tech Rpt Ser No 532. Geneva:World Health Organization, 1973.
49. ATSDR. Toxicological Profile for Manganese. Atlanta, GA:Agency for Toxic Substances and Disease Registry, 1996.
50. Hurley LS, Keen CL. Manganese. In: Trace Elements in Human and Animal Nutrition. Vol 1 (Mertz W, ed). Orlando, FL:Academic Press, 1987;185-223.
51. Hambidge KM, Casey CE, Krebs NF. Zinc. In: Trace Elements in Human and Animal Nutrition (Mertz W, ed). 5th ed. Orlando, FL:Academic Press, 1986;1-137.
52. Cunningham BC, Bass S, Fuh G, Wells JA. Zinc mediation of the binding of human growth hormone to the human prolactin receptor. Science 250:1709-1712 (1990).
53. Prasad AS, Brewer GJ, Schoomaker EB, Rabbani P. Hypocupremia induced by zinc therapy in adults. J Am Med Assoc 240:2166-2168 (1978).
54. Yadrick MK, Kenney MA, Winterfeldt EA. Iron, copper, and zinc status: response to supplementation with zinc or zinc and iron in adult females. J Am Clin Nutr 49:145-150 (1989).
55. Fisher PWF, Giroux A, L'Abbe M. Effect of zinc supplementation on copper status in adult man. Am J Clin Nutr 40:743-746 (1984).
56. Chandra RK. Excessive intake of zinc impairs immune responses. J Am Med Assoc 252:1443-1446 (1984).
57. Hooper PL, Visconti L, Garry PJ, Johnson GE. Zinc lowers high-density lipoprotein-cholesterol levels. J Am Med Assoc 244:1960-1961 (1980).
58. Prasad AS. Zinc in Human Nutrition. Boca Raton, FL:CRC Press, 1979.
59. Holt AB, Sprago RM, Iveson JB, Faulker GS, Cheek, DB. Serum and plasma zinc, copper and iron concentrations in aboriginal communities of northwestern Australia. Am J Clin Nutr 83:119-136 (1980).
60. Fraker PJ, Gershwin ME, Good RA, Prasad A. Interrelationships between zinc and immune function. Fed Proceed 45:1474-1479 (1986).
61. ATSDR. Toxicological Profile for Zinc. Atlanta, GA:Agency for Toxic Substances and Disease Registry, 1994.
62. Clewell HJ, Anderson ME. Risk assessment extrapolations and physiological modeling. Toxicol Ind Health 14:111-131 (1985).
63. ATSDR. Toxicological Profile for Methylene Chloride. Atlanta, GA:Agency for Toxic Substances and Disease Registry, 1993.
64. Winneke G. Behavioral effects of methylene chloride and carbon monoxide as assessed by sensory and psychomotor performance. In: Behavioral Toxicology (Xintaras C, Johnson BL, de Groot I, eds). Washington:U.S. Government Printing Office, 1974;130-144.
65. Clewell HJ. The use of physiologically based pharmacokinetic modeling in risk assessment: a case study with methylene chloride. In: Low-Dose Extrapolation of Cancer Risks: Issues and Perspectives (Farland W, Olin S, Park C, Rhomberg L, Sheuplin R, Starr T, Wilson J, eds). Washington:ILSI Press, 1995.
66. Fredericksson A, Danielsson BRG, Eriksson P. Altered behavior in adult mice orally exposed to tri- and tetrachloroethylene as neonates. Toxicol Lett 66:13-19 (1993).
67. Clewell HJ, Gentry PR, Gearhart JM, Allen BC. Evaluation of pharmacokinetic models for use in quantitative risk assessment: example with trichloroethylene. ICF Kaiser International report to OSHA/DHSP and EPA/OHEA, 1994.
68. U.S. EPA. The Use of the BMD Approach in Health Risk Assessment. EPA 603-R-94-007. Washington:U.S. Environmental Protection Agency, 1995.
69. Crump K. A new method for determining allowable daily intakes. Fundam Appl Toxicol 4:854-871 (1984).
70. Allen BC, Kavlock RJ, Kimmel CA, Faustman EM. Dose-response assessment for developmental toxicity. II: Comparison of generic BMD estimates with NOAELs. Fundam Appl Toxicol 23:487-495 (1994).
71. ATSDR. Toxicological Profile for Mercury. Atlanta, GA:Agency for Toxic Substances and Disease Registry, 1994.
72. McLachlan JA. Functional toxicology: a new approach to detect biologically active xenobiotics. Environ Health Perspect 101(5):386-387 (1993).
73. Mumtaz MM, Durkin PR. A weight-of-evidence approach for assessing interactions in chemical mixtures. Toxicol Ind Health 8(6):377-406 (1992).
74. Jonker D, Woutersen RA, van Bladeren PJ, Til HP, Feron VI. Subacute (4-week) oral toxicity of a combination of four nephrotoxins in rats: comparison with the toxicity of the individual compounds. Food Chem Toxicol 31(2):125-136 (1993).
75. Jonker D, Woutersen RA, Freon VJ. Toxicity of mixtures of nephrotoxicants with similar or dissimilar mode of action. Food Chem Toxicol 34:1075-1082 (1996).
76. Johnson BJ, Jones DE. ATSDR's activities and views on exposure assessment. J Expo Anal Environ Epidemiol 2 (Suppl 1):1-17 (1992).
Last Update: March 11, 1998