Toxicological
Defense Mechanisms and the Shape of Dose-Response Relationships
Environmental
Health Perspectives 106, Supplement 1, February 1998
ICF Kaiser Engineers, Incorporated, Research Triangle Park, North Carolina
Key words: exposure biomarkers, response biomarkers, dose-response relationship, 2,3,7,8-tetrachlorodibenzo-p-dioxin, TCDD, response curves, U-shaped curves, markers of tumor promotion
This paper is based on a presentation at The Third BELLE Conference on Toxicological Defense Mechanisms and the Shape of Dose-Response Relationships held 12-14 November 1996 in Research Triangle Park, NC. Manuscript received at EHP 7 March 1997; accepted 6 June 1997.
Address correspondence to Dr. M.E. Andersen, ICF Kaiser Engineers, Inc., P.O. Box 14348, Research Triangle Park, NC 27709. Telephone: (919) 547-1723. Fax: (919) 547-1710. E-mail: andersenme@aol.com
Abbreviations used: Ah, aryl hydrocarbon; arnt, aryl hydrocarbon nuclear transferase; DEN, diethylnitrosamine; DREs, dioxin response elements; GST, glutathione S-transferase; PCB, polychlorinated biphenyl; PCDD, polychlorinated dibenzo-p-dioxin; PH, partial hepatectomy; TCDD, 2,3,7,8-tetrachlorodibenzo-p-dioxin.
In these cases, accurate estimation of risks to human health posed by low-level exposure to potentially toxic compounds requires knowledge of the dose-response relationship over a broad range of exposures in the animal species. Unfortunately, dose-response curves that define risks in animals at low response rates are almost nonexistent and in some cases may be impossible to obtain. For toxic responses with measurable background rates, such as cancer and teratogenesis, it is difficult to assess the significance of small increases above background response rates. These studies only infrequently resolve differences of response in the 1 in 20 to 1 in 100 range. When evaluating impaired function such as reproductive competence, the natural variability in a healthy population of measures of effect such as numbers of live births per litter greatly restrict accurate assessment of low-level response rates related to chemical exposures.
It may be possible to extend dose-response curves to lower levels of exposure based on theoretical knowledge of the mechanisms of toxicity by using so-called biologically based models or by measuring precursor biochemical or molecular events directly involved in the sequence of events leading to toxicity states. Proposed revised carcinogen risk assessment guidelines (1) discuss the use of these precursor events to extend the dose-response curve to low levels of exposure and the role of biologically based models in cancer risk assessment. Precursor events should be causally related to toxicity to be useful in risk assessment. A precursor response linked mechanistically to toxicity is equivalent to a biomarker of response.
With DNA-reactive carcinogenic compounds, response biomarkers include the presence of chemical-related DNA adducts and the identification of specific mutations in oncogenes. With compounds that serve as tumor promoters, exposure may lead to altered regulation of growth regulatory genes or to recurrent toxicity with concomitant reparative hyperplasia. With either DNA-reactive or indirect-acting carcinogens, the various response biomarkers can be viewed in either qualitative or quantitative fashion.
If there is no evidence of an increase in response biomarkers, there should be no increase in expected risk from a toxic compound in target populations. This application provides a qualitative argument of the relationship between the biomarker and the presumed risk. However, based on the statistical power of the test measurement there is still some level of increase in the biomarker and therefore some level of increased risk that cannot be evaluated based directly on the measurements. A more difficult question involves the quantitative relationship between the intensity of the biomarker in the organism and the risk of overt toxicity. To use these precursor events in quantitative risk estimation for low-level exposures, it is necessary to understand the relationship between these precursor changes and toxic responses over the full dose range of interest. This paper, using experience with 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) as an example, examines the advantages and disadvantages of using response biomarkers to estimate dose-response relationships quantitatively at low levels of exposures.
Our understanding of the mechanisms of TCDD interactions with the Ah receptor are based primarily on studies of CYP1A1 induction by TCDD. The inactive Ah receptor consists of an aggregate of several proteins (8).
Binding TCDD to the Ah receptor aggregate leads to dissociation of the Ah receptor from the aggregate and dimerization of the Ah-TCDD complex with another protein, the aryl hydrocarbon nuclear transferase (arnt). Both the Ah protein (9) and arnt (10) are ß-helix-loop-helix DNA binding proteins. The heterodimer formed from these two proteins has a high affinity for specific DNA response elements found upstream from the CYP1A1 gene. For other proteins, such as CYP1A2, gene transcription is increased by the Ah-TCDD complex even though there is no CYP1A1-like response element upstream from this gene (11).
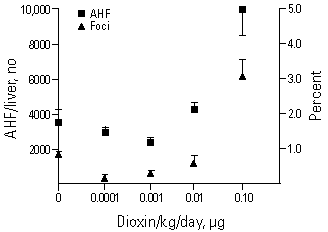
Figure 1. Effects of TCDD treatment on the growth of altered hepatic foci (AHF) in female rats. Rats were administered single doses of DEN as an initiating agent 24 following 70% PH. TCDD was dosed every 2 weeks to provide an average daily dose as specified on the x-axis. The AHF represent any and all foci for three markers---glutamyltranspeptidase, adenosine triphosphatase, and glucose 6-phosphatase. Data from Pitot et al. (12). The doses used correspond to the daily doses used in the original cancer bioassay (2). In the original bioassay (2) the incidence of cancer at the three doses--0.001, 0.01, and 0.1 µg/kg/day--were 1/86, 0/50, 2/50, and 11/49, respectively. In the Pitot et al. study (12) the control rats were sacrificed at 8 months, while the treated rats were sacrificed at 6 months (36). These differences diminish the U-shaped behavior for volume occupied by AHF; they are not expected to affect the overall shape of the curve for AHF numbers in the liver.
Van Bergelen et al. (13) found that TCDD treatment increased the concentrations of porphyrins in the liver and that this increase correlated well with the induction of CYP1A2. Mechanistically, this correlation may arise because the metabolism of uroporphyrinogen III to uroporphyrin III is catalyzed by CYP1A2 (14). TCDD also increases the proliferation rate of hepatocytes and induces toxicity, defined as cytoplasmic vacuolation, fatty changes, bile duct hyperplasia, and pigment in Kupfer cells (15). Stinchcombe et al. (16) found that apoptotic rates of the cells in altered foci staining positively for glutathione S-transferase (GST) produced by TCDD treatment were much lower than the apoptotic rates measured in these foci in the absence of TCDD. Any of these various processes may be regarded as precursor steps to the development of liver tumors.
The most widely used marker in experimental animals for exposure to PCDDs and similar compounds is the induction of the CYP1A family cytochromes. This sensitive biomarker is not specific to TCDD or even to halogenated polyaromatic compounds. Other PCDDs, chlorinated dibenzofurans, some PCBs, and some natural products such as polyaromatic hydrocarbons also induce these proteins. Within a human population, increased CYP1A1 is a measure of exposure to a wide variety of compounds that activate the Ah-receptor aggregate.
Bars and Elcombe (18) first noted that CYP1A enzyme induction by TCDD was heterogeneous over the liver acinus. Tritscher et al. (19) reported induction in livers of rats treated for 3 months. At lower doses (3.5 and 10.7 ng TCDD/kg/day) induction occurred in the centrilobular region and progressively moved outward to the mid-zonal and then the periportal areas as dose increased up to 125 ng/kg/day. In addition, when the immunohistochemically stained slides were evaluated closely, cells were found to be either fully induced or in a basal state, i.e., there was a sharp boundary between areas of induced cells and areas with no induced cells (18). Thus, TCDD appears to cause a shift in the state of hepatocytes from the basal noninduced phenotype to a dioxin-activated phenotype in which CYP1A1 and CYP1A2 are fully induced.
Physiologically based pharmacokinetic models for TCDD that included gene induction were developed in the late 1980s and early 1990s (20-24). These initial CYP1A family induction models did not consider heterogeneous induction in evaluating the dose response for this molecular marker of the effects of TCDD on the liver. These earlier models were successful in explaining the induction averaged over the whole liver but required alteration to simultaneously describe the nonlinear induction of CYP1A1 mRNA by TCDD (17).
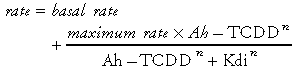 [1]
[1]
To account for regional induction with sharp boundaries, the presumed binding affinity of DREs for the Ah-TCDD complex (Kdi) were varied among five compartments in the liver. The five compartments (Figure 2) were defined geometrically as a hexagonal acinar structure (25). The model was capable of simulating available data on total induction of mRNA and CYP1A family proteins and on the regional patterns of induction when the Hill coefficients were large (4 or greater) and the Kdi values varied by a factor of 3 between compartments. This parameterization of the multicompartment liver model predicted a variation of 81 in the effective binding affinity over the five compartments.
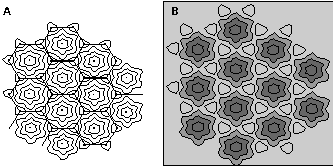
Figure 2. A schematic representation of the surfaces of hexagonal acinar structures within the liver in the geometric liver model used to predict regional induction characteristics with TCDD. The most recent physiologically based pharmacokinetic (PBPK) models for TCDD subdivide the liver acinus into five compartments based on concentric regions within the acinus (25,26). This geometric model (A) allows prediction of regional induction (B) by assuming different binding characteristics of TCDD with DNA response elements in each region. Estimates of total induction in a region are estimated and compared to maximally induced levels. The proportion induction is then used to calculate a color intensity for the region (B). To obtain sharp boundaries with some fully induced regions (the two centrilobular zones) and other noninduced regions (the two periportal zones), the Hill coefficients in the induction equations in the PBPK models must be 4 or greater. Any comprehensive model of hepatic induction by most CYP450 inducers should account for regional effects as well as effects averaged over the entire liver.
The requirement of a large n-value indicates a steep, highly nonlinear
dose-response relationship for induction in both the observed and low-dose
regions. In the published analysis by Andersen et al. (26) the dissociation
constant in the centrilobular region (the most sensitive region of the
liver for CYP1A induction) was estimated to be 0.11 nM Ah-TCDD for induction
of CYP1A2. With the n-value equal to 4, the proportionate response for
CYP induction in the centrilobular region at a 10-fold lower concentration
of Ah-TCDD complex (0.011 nM) would be reduced to 1![]() 10-4.
The response rate for the centrilobular hepatocytes would fall to one dioxin-activated
cell per 1,000,000 normal cells when the concentration of the Ah-TCDD complex
in this compartment is the Ah-TCDD complex-DNA dissociation constant divided
by 31.
10-4.
The response rate for the centrilobular hepatocytes would fall to one dioxin-activated
cell per 1,000,000 normal cells when the concentration of the Ah-TCDD complex
in this compartment is the Ah-TCDD complex-DNA dissociation constant divided
by 31.
Thus, risks associated with events that were directly dependent on induction and activation of cells to a dioxin-responding phenotype as obligate precursor event would be minimal (1 in 106) if the receptor complex concentration fell below 0.000355 nM. Similar conclusions regarding receptor-ligand concentrations associated with minimal risks would also be valid for other hepatic enzyme inducers with heterogeneous patterns of induction in which individual cells appear to be either in the normal state or fully induced. These compounds include phenobarbital, peroxisomal proliferators, and a variety of dioxinlike compounds, including dibenzofurans and coplanar PCBs.
The hepatocarcinogenicity of dioxin appears instead to result from the spontaneous appearance of mutated cells that have reduced responsiveness to the growth regulatory control affecting normal cells. In the presence of TCDD, mutated cells grow to be identifiable clones. As the clones increase in size, the opportunities for subsequent mutations to more aggressive cell types also increase. If the probability of mutation or the cell proliferation rates were simple functions of the cells recruited into the dioxin-activated state, it would be possible to estimate the dose-response curve for all levels of exposure from measurements of CYP1A1 induction. The evidence below, however, suggests that no simple relationship exists between carcinogenicity and the extent of enzyme induction.
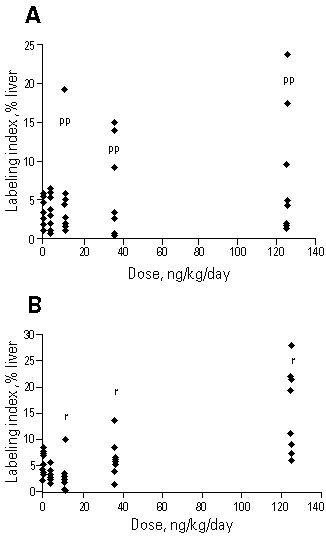
Figure 3. Labeling index in normal hepatocytes in female Sprague-Dawley rats at the end of a 90-day initiation/promotion study. (A) The rats in this study were the initiation controls. They were treated with TCDD but not initiated with DEN. In these rats labeling was not homogeneously observed throughout the acinus. At 10, 35, and 125 ng TCDD/kg/day, labeling was reported predominantly in the periportal (pp) area. (B) In this study rats were initiated with a necrotizing dose of DEN (175 mg/kg). In these rats labeling occurred randomly (r) throughout the liver acinus. The r and pp notations at the higher doses indicate that the majority of animals in these groups had proliferation reported as r or pp. Data from Maronpot et al. (15).
Bauman et al. (28) studied the effect of TCDD on the regeneration of liver after PH. Twenty-four hours after PH, 61% of the hepatocytes in control animals were in the cell cycle, i.e., cells identified by staining techniques to be in either G1, S, G2, or M phase of the cell cycle. With TCDD only 41% of the hepatocytes were in the cell cycle at a comparable time after PH. These results indicate that one of the functional responses of the liver to TCDD is a decrease in responsiveness of hepatocytes to mitogenic stimulation. The authors also found a decreased response of TCDD-treated livers to mitostimulation by lead nitrate. In general, the body of research with TCDD indicated mitosuppression and shift of proliferation to the periportal region at low doses, whereas cell proliferation throughout the liver and toxicity occur at higher doses.
In contrast, Andersen et al. (33) and Conolly and Andersen (34) evaluated
an alternative model for interpreting these initiation-promotion results
with TCDD. In their model DEN initiation produces two cell types (A and
B) capable of becoming clones of enzyme-altered foci in the livers. One
of these cell types, A, responds to the negative growth environment associated
with TCDD treatment. The net growth rate for this cell type, given by the
birth rate minus the death rate (![]() A-ßA),
decreases with increasing TCDD exposure. Most clones observed in DEN controls
in the absence of TCDD treatment are expected to be derived from these
A cells. The second type of cell, B, is unresponsive to the mitoinhibitory
environment associated with TCDD treatment. For these cells
A-ßA),
decreases with increasing TCDD exposure. Most clones observed in DEN controls
in the absence of TCDD treatment are expected to be derived from these
A cells. The second type of cell, B, is unresponsive to the mitoinhibitory
environment associated with TCDD treatment. For these cells ![]() B-ßB
increases with increasing concentrations of TCDD. At the high exposure
concentrations in the initiation-promotion studies, the observed clones
are derived primarily from these B cells. This two-cell model is consistent
with the observed U-shaped dose-response curves, explains TCDD promotion
without assuming a mutational component to the formation of the clones
over time, and is consistent with a mitoinhibitory action of TCDD on normal
cells that is absent in the B-cell clones (34).
B-ßB
increases with increasing concentrations of TCDD. At the high exposure
concentrations in the initiation-promotion studies, the observed clones
are derived primarily from these B cells. This two-cell model is consistent
with the observed U-shaped dose-response curves, explains TCDD promotion
without assuming a mutational component to the formation of the clones
over time, and is consistent with a mitoinhibitory action of TCDD on normal
cells that is absent in the B-cell clones (34).

Based on the gene induction model, we calculated the Ah-TCDD concentration
expected to be associated with a 1 in 106 level of induction
in the centrilobular compartment (3.5![]() 10-4
nM). Because the dose response for carcinogenicity is expected to be nonlinear
with contributions from multiple factors, the biomarker dose-response curve
at low levels of exposure would provide a conservative estimate of risk.
When the biomarker falls to an expected response level of 1 per 106,
the carcinogenic risk would be expected to be much below this level. Although
molecular parameters may be poor predictors of actual risk, these markers
could be used to provide a conservative bound. This more limited quantitative
use of the biomarker, although perhaps disappointing from a mechanistic
perspective, at least avoids low-dose extrapolation based on nothing more
than policy.
10-4
nM). Because the dose response for carcinogenicity is expected to be nonlinear
with contributions from multiple factors, the biomarker dose-response curve
at low levels of exposure would provide a conservative estimate of risk.
When the biomarker falls to an expected response level of 1 per 106,
the carcinogenic risk would be expected to be much below this level. Although
molecular parameters may be poor predictors of actual risk, these markers
could be used to provide a conservative bound. This more limited quantitative
use of the biomarker, although perhaps disappointing from a mechanistic
perspective, at least avoids low-dose extrapolation based on nothing more
than policy.
Several lines of evidence support U-shaped dose-response curves for hepatic effects of TCDD, including initiation/promotion (12), carcinogenicity (2), and cell labeling (15). The downward sloping portion of these curves appears to be in the range where mitosuppression occurs. This mitoinhibition appears to protect the liver from carcinoma production. It would be misleading, however, to call this low-dose protection.
The overall U-shaped curve appears to be associated with regions of the dose-response regime in which different effects of dioxin predominate over others. At high doses the combination of proliferation, toxicity, and mitoinhibition acts to enhance carcinogenicity. At lower doses mitoinhibition, acting in the absence of toxicity and proliferative responses, appears to moderate cell proliferation and reduce the incidence of foci production and tumors relative to those in controls. This region is not a dose region where all effects of TCDD are expected to be beneficial. It is a region in which the mitoinhibition in liver predominates over other effects. In other tissues the TCDD-related effects associated with these altered cell growth characteristics may be associated with other toxic effects. One caution from this evaluation of the possibility of U-shaped curves with TCDD relates to the definitions of both hormesis and low levels of exposure. Low levels of exposure, in this context, simply refer to levels below those that have overt increases in toxic responses. The dose that causes a low-level response in one tissue may be associated with much higher levels of response in a second tissue. Hormesis is an empirical definition based on observing complex dose-response curves for specific toxic responses. When we realize that the pathogenicities of most toxic responses are composites of multiple effects of the chemical on the organ system, it is easy to see that complex curves may arise from differential dose-response relationships for the contributing mechanisms of pathogenesis.
Biochemical and molecular parameters that are specific and sensitive may be useful for identifying doses below which increases in biomarker are not statistically significant. Experimental measurements alone cannot unequivocally establish a total lack of response because of the statistical power of the test systems. Biologically based pharmacodynamic models such as that for regional and cell-specific induction with TCDD have the potential to provide characterization of the functional relationships between dose and the biomarker and may increase our confidence in extending the predictions of the dose-response relationship for the biomarker to lower doses based on mechanistic considerations. In contrast to toxicity processes, molecular responses such as CYP1A family induction are simpler and depend on fewer biologic factors than toxic and carcinogenic sequelae. Therefore, these models of low-level increases in biomarkers are more likely to be testable by experimental studies than pharmacodynamic models of toxic responses. Despite difficulties in equating biomarker concentrations with specific degrees of risk, estimation of low-level risks based on these biomarkers is likely to be important in placing an upper bound on risk if it is assumed that a direct relation exists between the biomarker and toxicity.
References
1. U.S. EPA. Proposed guidelines for carcinogen risk assessment. EPA 600-P-92-003C. Washington:U.S. Environmental Protection Agency Office of Research and Development, 1996.
2. Kociba RJ, Keyes DG, Beyer JE, Carreon RM, Wade CE, Dittenber DA, Kalnins RP, Frauson LE, Park CN, Banard SD et al. Results of a two-year chronic toxicity and oncogenicity study of 2,3,7,8-tetrachlorodibenzo-p-dioxin in rats. Toxicol Appl Pharmacol 120:138-154(1978).
3. U.S. NTP. Carcinogenesis Bioassay of 2,3,7,8-Tetrachlorodibenzo-p-dioxin (CAS No. 1746-01-6) in Osborne-Mendel Rats and B6C3F1 Mice (Gavage Study). Tech Rpt No 209. Research Triangle Park, NC:U.S. National Toxicology Program, 1982.
4. Abbott BD, Birnbaum LS, Pratt RM. 2,3,7,8-TCDD-induced hyperplasia of the ureteral epithelium produces hydronephrosis in murine fetuses. Teratology 35:3219-3233(1987).
5. Burelson GR, Lebrec H, Yang YG, Ibanes JD, Pennington KN, Birnbaum LS. Effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on influenze virus host resistance in mice. Fundam Appl Toxicol 29:40-47 (1996).
6. Mably TA, Bjerke DL, Moore RW, Gendron-Fitzpatric A, Person RE. in utero and lactational exposure of male rats to 2,3,7,8-tetrachlorodibenzo-p-dioxin. 3: Effects of spermatogenesis and reproductive capability. Toxicol Appl Pharmacol 114:118-128 (1993).
7. Gray LE Jr, Kelce WR, Monosson E, Ostby JS, Birnbaum LS. Exposure to TCDD during development permanently alters reproductive function in male Long Evans rats and hamsters: reduced ejaculated and epididymal sperm numbers and sex accessory gland weights in offspring with normal androgenic status. Toxicol Appl Pharmacol 131:108-118 (1995).
8. Perdew G. Chemical cross-linking of the cytosolic and nuclear forms of the Ah receptor in hepatoma cell line 1C1C7. Biochem Biophys Res Comm 182:55-63 (1992).
9. Swanson HI, Bradfield CA. The Ah-receptor: genetics, structure and function. Pharmacogenetics 3:213-230 (1993).
10. Reyes H, Reisz-Porszasz S, Hankinson O. Identification of the Ah receptor nuclear translocator protein (arnt) as a component of the DNA binding form of the Ah receptor. Science 256:1193-1195 (1992).
11. Quattrochi LC, Vu T, Tukey RH. The human CYP1A2 gene and induction by 3-methylcholanthrene. A region of DNA supoorts Ah-receptor binding and promoter-specific induction. J Biol Chem 269:6949-6954 (1994).
12. Pitot H, Goldsworthy TL, Moran S, Kennan W, Glauert HP, Maronpot RR, Campbell HA. A method to quantitate the relative initiating and promoting potencies of hepatocarcinogenic agents in their dose-response relationships to altered hepatic foci. Carcinogenesis 8:1491-1499 (1987).
13. van Birgelen APJM, DeVito MJ, Akins JM, Ross DG, Diliberto JJ, Birnbaum LS. Relative potencies of polychlorinated dibenzo-p-dioxins, dibenzofurans and biphenyls derived from hepatic porphyrin accumulation in mice. Toxicol Appl Pharmacol 138:98-109 (1996).
14. Jacobs JM, Sinclair PR, Bement WJ, Lambrecht RW, Sinclair JF, Goldstein JA. Oxidation of uroprophyrinogen by methycholanthrene-induced cytochrome P-450. Essential role of cytochrome P450d. Biochem J 258:247-253 (1989).
15. Maronpot RR, Foley JF, Takahashi K, Goldsworthy T, Clark G, Trischer A, Portier C, Lucier G. Dose response for TCDD promotion of hepatocarcinogenesis in rats initiated with DEN: histologic, biochemical and cell proliferation end points. Environ Health Perspect 101:634-642 (1993).
16. Stinchcombe S, Buchmann A, Bock KW, Schwarz M. Inhibition of apoptosis during 2,3,7,8-tetrachlorodibenzo-p-dioxin-mediated tumour promotion in rat liver. Carcinogenesis 16:1271-1275 (1995).
17. Vanden Heuvel JP, Clark GC, Kohn MC, Tritscher AM, Greenlee WF, Lucier GW, Bell DA. Dioxin-responsive genes: examination of dose-response relationships using quantitative reverse transcriptase-polymerase chain reaction. Cancer Res 54:62-68 (1994).
18. Bars RG, Elcombe CR. Dose-dependent acinar induction of cytochromes P450 in rat liver: evidence for a differential mechanism of induction of P450IA1 by ß-naphthoflavone and dioxin. Biochem J 277:577-580 (1991).
19. Tritscher M, Goldstein JA, Portier CJ, McCoy Z, Clark GC, Lucier GW. Dose-response relationships for chronic exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin in a rat tumor promotion model: quantification and immunolocalization of CYP1A1 and CYP1A2 in the liver. Cancer Res 52:3436-3442 (1992).
20. Leung HW, Ku RH, Paustenbach DJ, Andersen ME. A physiologically based pharmacokinetic model for 2,3,7,8-tetrachloro-p-dioxin in C57B1/6J and DBA/2J mice. Toxicol Lett 42:15-28 (1988).
21. Leung HW, Paustenbach DJ, Murray FJ, Andersen ME. A physiological pharmacokinetic description of the tissue distribution and enzyme inducing properties of 2,3,7,8-tetrachlorodibenzo-p-dioxin in the rat. Toxicol Appl Pharmacol 103:399-410 (1990).
22. Kohn MC, Lucier GW, Clark GC, Sewall C, Tritscher A, Portier CJ. A mechanistic model of the effects of dioxin on gene expression in the rat liver. Toxicol Appl Pharmacol 120:138-154 (1993).
23. Kedderis LB, Mills JJ, Andersen ME, Birnbaum LS. A physiologically based pharmacokinetic model for 2,3,7,8-tetrabromodibenzo-p-dioxin (TBDD) in the rat: tissue distribution and CYP1A induction. Toxicol Appl Pharmacol 121:87-98 (1993).
24. Andersen ME, Mills JJ, Gargas ML, Kedderis L, Birnbaum LS, Neubert D, Greenlee WF. Modeling receptor-mediated processes with dioxin: implications for pharmacokinetics and risk assessment. J Risk Anal 13:25-36 (1993).
25. Andersen ME, Eklund CR, Mills JJ, Barton HA, Birnbaum LS. A multi-compartment geometric model of the liver in relation to regional induction of cytochrome P450s. Toxicol Appl Pharmacol 144:135-144 (1997).
26. Andersen ME, Birnbaum LS, Barton HA, Eklund CR. Regional hepatic CYP1A1 and CYP1A2 induction with 2,3,7,8-tetrachlorodibenzo-p-dioxin evaluated with a multicompartment geometric model of hepatic zonation. Toxicol Appl Pharmacol 144:145-155 (1997).
27. Fox TR, Best LL, Goldsworthy SM, Mills JJ, Goldsworthy TL. Gene expression and cell proliferation in rat liver after 2,3,7,8-tetrachlorodibenzo-p-dioxin exposure. Cancer Res 53:2265-2271 (1993).
28. Bauman JW, Goldsworthy TL, Dunn CS, Fox TR. Inhibitory effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin on rat hepatocyte proliferation induced by 2/3 partial hepatectomy. Cell Prolif 28:437-451 (1995).
29. Portier CJ, Kohn MC, Kopp-Schneider A, Sherman CD, Maronpot R, Lucier GW. Modeling the number and size of hepatic focal lesions following exposure to 2,3,7,8-TCDD. Toxicol Appl Pharmacol 138:20-30 (1996).
30. Moolgavkar SH, Luebeck EG, Buchmann A, Bock KW. Quantitative analysis of enzyme-altered liver foci in rats initiated with diethylnitrosamine and promoted with 2,3,7,8-tetrachlorodibenzo-p-dioxin or 1,2,3,4,6,7,8-heptachlorodibenzo-p-dioxin. Toxicol Appl Pharmacol 138:31-42 (1996).
31. Rogers A, Andersen ME, Back KC. Mutagenicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin and perfluorodecanoic acid LS178Y mouse lymphoma cells. Mutat Res 105:445-449 (1982).
32. Yang J-H, Thraves P, Dritschilo A, Rhim JS. Neoplastic transformation of immortalized human keratinocytes by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Cancer Res 52:3478-3482 (1992).
33. Andersen ME, Mills JJ, Jirtle RL, Greenlee WF. Negative selection in hepatic tumor promotion in relation to cancer risk assessment. Toxicology 102:223-237 (1995).
34. Conolly RB, Andersen ME. Hepatic foci in rats after diethylnitrosamine initiation and 2,3,7,8-tetrachlorodibenzo-p-dioxin promotion: evaluation of a quantitative two-cell model and of CYP1A1/1A2 as a dosimeter. Toxicol Appl Pharmacol 146:281-293 (1997).
35. Jirtle RL, Meyer SA, Brockenbrough JS. Liver tumor promoter phenobarbital: a biphasic modulator of hepatocyte proliferation. In: Chemically Induced Cell Proliferation: Implications for Risk Assessment (Butterworth B, Slaga TJ, Farland W, McClain M, eds). Vol 369. New York:Wiley-Liss, 1991; 209-216.
36. Pitot H. Personal communication.
Last Update: March 11, 1998